
機能詳細
第一原理電子状態計算プログラム MedeA-VASP
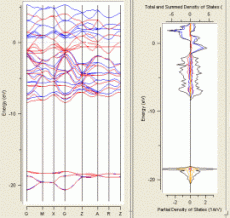 MedeAは、第一原理電子状態計算プログラムとして、VASP(*1)を搭載しています。VASP は、密度汎関数法を用いた平面波- 擬ポテンシャル法電子状態計算プログラムです。Blöchl のProjector Augmented Wave(PAW)法によるポテンシャルライブラリを実装しており、高速で精度の高い計算を実現します。安定構造や遷移状態構造の最適化、拡散パスの検討、反応解析など様々な問題に適用されています。
MedeAは、第一原理電子状態計算プログラムとして、VASP(*1)を搭載しています。VASP は、密度汎関数法を用いた平面波- 擬ポテンシャル法電子状態計算プログラムです。Blöchl のProjector Augmented Wave(PAW)法によるポテンシャルライブラリを実装しており、高速で精度の高い計算を実現します。安定構造や遷移状態構造の最適化、拡散パスの検討、反応解析など様々な問題に適用されています。
1. VASP(Vienna Ab initio Simulation Package)はウィーン大学で開発された第一原理電子状態計算プログラムです。
G. Kresse and J. Furthmüler, Phys. Rev. B 54, 11169 (1996); Computat. Mat. Sci. 6, 15 (1996).
G. Kresse and D. Joubert, Phys. Rev. B 59, 1758 (1999).
 機能
機能
- 内殻電子の取扱い
PAWポテンシャル - 汎関数
密度汎関数
Van der Waals 密度汎関数(optB86b-vdW等)
Meta-GGA(revTPSS, TPSS, M06-L, MBJLDA)
ハイブリッド汎関数(PBE0, HSE06, B3LYP)
Screened exchange
Hartree-Fock - 交換相関ポテンシャル
局所密度近似(LDA)
一般化密度勾配補正(GGA-PBE, GGA-PBEsol等) - 分散力補正(GrimmeのDFT-D2)
DFT-D3, DFT-D2(Grimme)
Tkatchenko-Scheffler - 局在クーロン相互作用(LDA+U)
- Monkhorst-Pack の特殊点法によるk 点の自動発生
- Brillouin 領域での積分と占有電子の取扱い
Tetrahedron 法(Blöchl の補正)
Smearing(Gaussian、Fermi、Methfessel-Paxton) - スピン分極、スピン軌道相互作用、ノンコリニア磁性
- GW近似
- 第一原理分子動力学計算
ミクロカノニカルアンサンブル (NVE)
カノニカルアンサンブル (NVT)
等温等圧アンサンブル (NPT) - 溶媒効果(Implicit model)
物性
- 全エネルギー、生成エネルギー、
力、応力テンソル - 格子定数と原子座標の同時緩和
- バンド構造
- 状態密度 (全状態密度、部分状態密度)
- 電荷密度、静電ポテンシャル
- スピン密度
- ELF (Electron Localization Function)
- 原子座標、静電場の変化に対する線型応答
誘電テンソル、ピエゾテンソル、
ボルン有効電荷テンソル - 弾性テンソル
- 光学スペクトル
誘電関数、屈折率、吸収係数、伝導率 - 超微細構造定数
- NMR化学シフト
VASP GUI
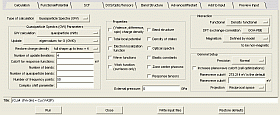 構築した構造モデルに対応した入力ファイルを自動的に作成します。VASPインターフェース上で、パラメータを変更することができ、対応したk点やポテンシャルファイルを自動的に作成します。設定されたジョブは、MedeA独自のキューイングシステム(Job Server / Task Server)と連携し、リモートマシンに投入することができます。
構築した構造モデルに対応した入力ファイルを自動的に作成します。VASPインターフェース上で、パラメータを変更することができ、対応したk点やポテンシャルファイルを自動的に作成します。設定されたジョブは、MedeA独自のキューイングシステム(Job Server / Task Server)と連携し、リモートマシンに投入することができます。
自動収束テスト機能 Automated Convergence
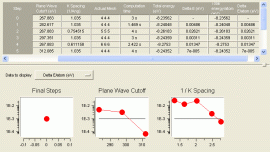 VASP で使用するパラメータは、その収束状況を判断して、最適なパラメータの組合せを設定する必要があります。Automated Convergence では、主なパラメータ(カットオフ値、k 点数、スメアリング手法)について、エネルギーに対する収束状況を自動的に計算し、最適なパラメータの組合せを求めます。
VASP で使用するパラメータは、その収束状況を判断して、最適なパラメータの組合せを設定する必要があります。Automated Convergence では、主なパラメータ(カットオフ値、k 点数、スメアリング手法)について、エネルギーに対する収束状況を自動的に計算し、最適なパラメータの組合せを求めます。
MedeA-VASPと組み合わせ可能な物性推算ツール
- 格子振動特性・熱力学物性評価ツール MedeA-Phonon
- 弾性特性・熱力学物性評価ツール MedeA-MT
- 電子状態解析ツール MedeA-Electronics
- クラスタ展開法ツール MedeA-UNCLE
- 遷移状態探索ツール Transition State Search
- SciMAPS
- 材料設計支援プラットフォーム