


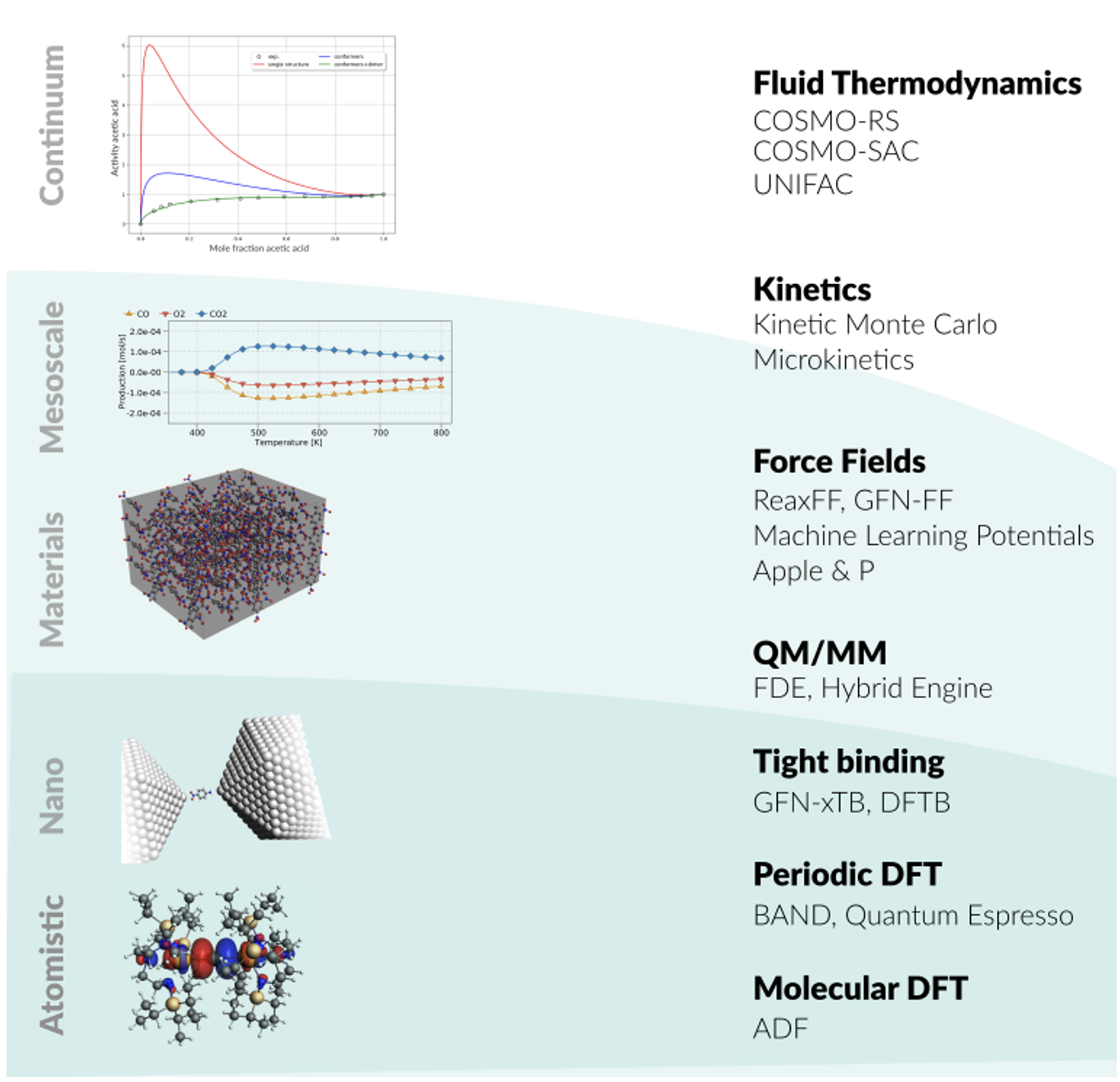
AMS
(Amsterdam Modeling Suite)
tab:{
title:{概要}
概要
AMS -Amsterdam Modeling Suite-
AMS(Amsterdam Modeling Suite)は、量子化学計算プログラムのADFをはじめとする計算化学統合パッケージです。
ADF (Amsterdam Density Functional)は、オランダAmsterdam自由大学のBaerends教授とカナダCalgary大学のZiegler教授の両グループが中心になって開発された、30年以上の歴史を誇る密度汎関数法 (DFT) を用いた量子化学計算プログラムです。
AMS (Amsterdam Modeling Suite)は下記製品群からなります。
・分子系のDFT計算プログラムADF & ADF-GUI
・周期系のDFT計算プログラムBAND & BAND-GUI
・熱力学物性計算プログラムCOSMO-RS
・反応分子動力学計算プログラムReaxFF
・密度汎関数タイトバインディング計算プログラムDFTB
ADFとBANDは、周期律表の全元素に対応した基底関数を完備しており、気相中、溶液中、タンパク質、金属表面上など、様々な環境下での計算に対応しています。ADFは、以下の特長を持ち、幅広い分野の研究に利用されています。 ・豊富なプロパティ計算機能(IR, NMR, ESR, UV/Vis, CD, etc.)
・遷移金属錯体での安定したSCFの収束 相対論効果の考慮(Scalar, Spin-orbit)
・全電子計算(擬ポテンシャル・ECPを使用しない)
・Slater型軌道による原子基底関数

ニュースレター
MOLSISニュースレターに掲載した記事を電子ブックで閲覧できます。
開発元SCM社について
Software for Chemistry & Materials BV(SCM社)は、1995年にBaerendsグループのスピンオフ企業として発足した会社で、AMSの開発、保守、販売において主導的な役割を担っています。SCM社のスタッフは、理論化学・理論物理分野でのPh.D.を持ち、AMSの開発やアプリケーションに長い経験をもつ技術者で構成されています。
AMS開発元 SCM社ウェブサイト もあわせてご覧ください。

title:{製品構成}
製品一覧
AMS (Amsterdam Modeling Suite)は以下の製品群で構成されます:
・分子系DFT:ADF & ADF-GUI
・周期系DFT:BAND & BAND-GUI
・熱力学物性:COSMO-RS
・反応分子動力学:ReaxFF
・高速タイトバインディングDFT:DFTB
・機械学習ポテンシャルと古典力場:MLPot & FF
・先進的ワークフローツール群:Advanced Workflows※
※ParAMS(パラメータ作成支援)、ChemTraYzer2(反応トラジェクトリ解析)、OLED workflows(有機EL材料評価)、ACE-Reaction(反応経路探索)、MKMCXX(マイクロキネティクス)、Zacros(速度論的モンテカルロ)
ADF
様々な系に適用可能な密度汎関数法ソフトウェア
Amsterdam Density Functional (ADF)は、密度汎関数法(DFT)に基づく量子化学計算ソフトウェアで、均一系、不均一系触媒から無機化学、重元素化学、生化学、各種分光学まで幅広い分野の研究に利用されています。DFTは、1990年代初頭から広く使われるようになった方法で、電子相関を含んでいる点でHartree-Fock法よりも高精度です。また、post Hartree-Fock法(MP2, CI, CCなど)では取り扱いが困難なサイズである数百原子からなる化合物についても高速に計算することができます。ADFは遷移金属を含んだ系でもSCF計算の収束がよく、重元素系で重要な相対論の効果を含めることができます。さらに、周期表の全元素に対応した全電子計算用の基底関数(Slater型)を完備していることは、ADFの大きな特長の一つです。
豊富なプロパティ計算機能
ADFは種々の分子物性を高精度に計算することができます。時間依存密度汎関数法(TD-DFT)により、紫外・可視吸収スペクトル(振動子強度と一重項・三重項励起エネルギー)や、周波数依存分極率、ラマン強度、ファン・デル・ワールス分散係数などを計算します。また、IRの振動数や強度、多重極子モーメントと同様に、NMRの化学シフトやスピン-スピンカップリング、ESR(EPR) g-テンソル、磁気的・電気的超微細構造テンソル、核四重極子カップリング定数の計算が可能です。ほとんどの物性計算に対して相対論の効果を考慮した計算を行うことができます。
計算方法
・(post) Hartree-Fock法: RHF, UHF, MP2
・密度汎関数法(Kohn-Sham法)
LDA, Xα
GGA: BP, PW91, mPW, BLYP, PBE, RPBE, revPBE, mPBE, OLYP, OPBE, etc.
Hybrid: B3LYP, O3LYP, BHandHLYP, B1PW91, MPW1PW, MPW1K, PBE0, OPBE0, etc.
(hybrid) meta-GGA: KCIS, VS98, FT97, BLAP3, BOP, OLAP3, TPSS, TPSSh, B97, M05, M06, etc.
Range Separated: LCY-BLYP, CAM-B3LYP, HSE06, ωB97X, etc.
Double Hybrid: rev-DOD-PBEP86-D4,rev-DOD-BLYP-D4, B2PLYP-D3BJ, B2KPLYP, etc.
MM Dispersion corrected: BLYP-D, PBE-D, BP86-D, TPSS-D, B3LYP-D, B97-D, etc.
Model: SAOP, GRAC, LB94
・post Kohn-Sham法: RPA, GW
・相対論効果: Scalar (ZORA), Spin-Orbit (ZORA)
・励起状態: Time Dpendent DFT, GW-BSE
・溶媒効果: COSMO, DRF, SCRF, 3D-RISM, DFT/DFT
・外場の設定: 点電荷、静電場、non-self-consistent Green’s function計算
・Constrained DFT: 電荷とスピンの拘束に対応
・QM/MM: IMOMM/ADF, AddRemove, QUILD
・分子動力学計算: マルチスケールMD, Biased MD(メタダイナクミス、アンブレラサンプリングなど)
構造予測・反応解析
構造最適化計算: quasi-Newton
遷移状態探索: EF, NEB
反応経路: Linear Transit, IRC
プロパティ計算
・熱力学物性: エントロピー、モル比熱など
・IRスペクトル、(共鳴)ラマンスペクトル、振動ラマン光学活性(VROA)、Franck-Condon因子
・紫外・可視吸収スペクトル、X線吸収スペクトル
・りん光発光:輻射速度、ゼロ磁場分裂
・円二色性スペクトル(VCD, ECD)、旋光分散スペクトル(ORD)
・磁気円二色性スペクトル(MCD)、Verdet定数、Faraday A, B項
・コアイオン化ポテンシャル(XPS)
・NMR: 化学シフト、スピン-スピンカップリング
・ESR: gテンソル、Aテンソル、ZFS、NQCC(EFG)、Qテンソル
・メスバウアースペクトル、NRVSスペクトル
・電気的多重極子モーメント、(周波数依存)分極率・超分極率、(動的)磁化率
・ファン・デル・ワールス分散係数
・原子電荷: Mulliken, Hirshfeld, Voronoi, AIM
・ホッピング伝導: 電荷移動積分(charge transfer integral)に基づく分子間の輸送評価
・励起状態間: スピン軌道相互作用行列要素(SOCME)、遷移双極子モーメント(TDM)
・励起子移動積分:ROSE、FOCDFT
解析機能
・フラグメント軌道による相互作用解析: フラグメントはユーザ定義が可能
・結合エネルギー解析、ETS-NOCV
・結合次数解析: Mayer, Nalewajski-Mrozek
精度・効率
・Slater型軌道による原子基底関数:周期表の全元素に対応した全電子基底関数およびFrozen Core基底関数を完備
・電子密度フィッティングによるクーロン相互作用の高速化
・基底関数のカットオフ: O(N)
ADF-GUI
ADF-GUIは、ADF用のグラフィカルユーザインターフェースです。ADF-GUIの各モジュールは有機的に連携しており、関連のある計算結果を簡単に表示することができます。例えば、ADFspectraでIRスペクトルを表示しながら、特定のピークを指定することでADFmovieに振動モードのアニメーションを表示させることができます。
構造構築機能
環構造・グループなどのフラグメントを用いた構造構築
分子力場・外部プログラム(DFTB, MOPAC)を使用した簡易構造最適化
フラグメントライブラリのカスタマイズ
表示機能
エネルギー準位図、軌道相関図の表示
3次元データの等値面・等高線表示: 分子軌道、電荷密度、静電ポテンシャルなど
各種スペクトルのグラフ表示: IR、ラマン、UV/Vis、CD、NMRなど
アニメーション表示: 構造最適化計算、基準振動解析、IRC計算
計算設定・ジョブ管理機能
計算方法、基底関数、プロパティなどの計算設定
計算ジョブの投入・管理機能
複数ジョブの作成・解析機能
BAND
様々な系に適用可能な高精度バンド計算ソフトウェア
BANDは、ポリマー、表面、バルク結晶などの研究を目的とした周期系を取り扱う密度汎関数法ソフトウェアです。擬ポテンシャル近似は用いず、全電子計算またはFrozen Core近似を使用した高精度な計算が可能です。BANDは、平面波を基底関数として採用する多くのバンド計算プログラムとは異なり、1〜2次元の周期境界条件を課すことが可能で、真のスラブ近似を使用した効率的な計算が可能です。基底関数には原子軌道(数値型+Slater型)を採用しており、周期表の全元素に対応した基底関数を完備しています。また、姉妹プログラムであるADFと同じ相対論法を搭載しているため、重原子を含む様々な系に適用可能です。
原子軌道の特徴を活かしたプロパティ計算・解析機能
BANDは、基底関数として原子軌道を採用しているため、部分状態密度や原子電荷など、原子に帰属させるプロパティの計算に適しています。不均一触媒の研究にしばしば使用され、金属表面への化学吸着や化学反応のポテンシャルエネルギー曲面の解析に応用されています。BANDでは、様々な交換相関汎関数が使用でき、結合(凝集)エネルギー解析、Mullikenポピュレーション解析、電荷密度フーリエ解析(構造因子)が可能です。また、フラグメント解析機能の特徴から、各フラグメントの分子軌道を用いた状態密度データの分割を行うことができます。さらに、時間依存密度汎関数法(TD-DFT)による精度の高い周波数依存誘電関数の計算や、周期系に対するNMR・ESR計算が可能です。
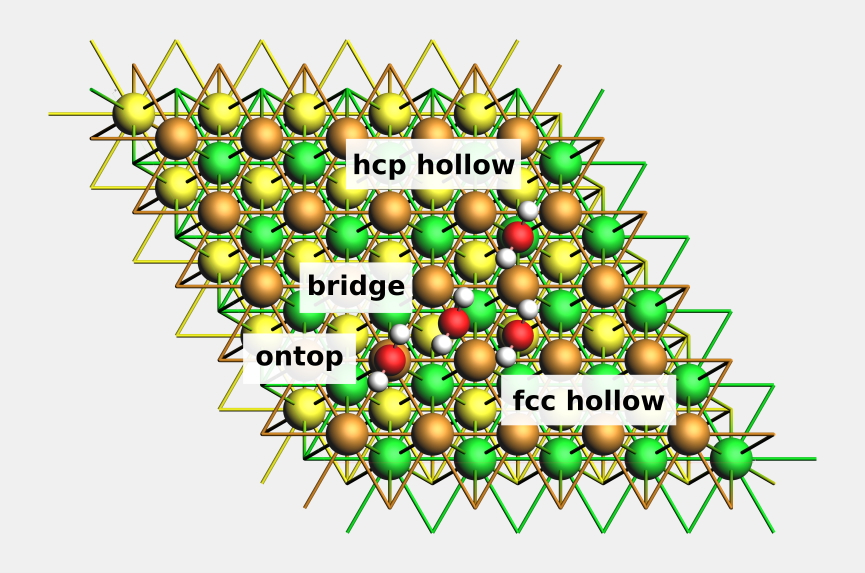
計算方法
密度汎関数法: LDA, GGA(BP, PW91, BLYP, PBE, PBEsol, etc.), GGA-D(D, D3, D3(BJ)), Meta-GGA(MO6L, TPSS, etc.), model(LB94, TB-mBJ), LDA+U
相対論効果: Scalar(ZORA), Spin-Orbit(ZORA)
溶媒効果: COSMO
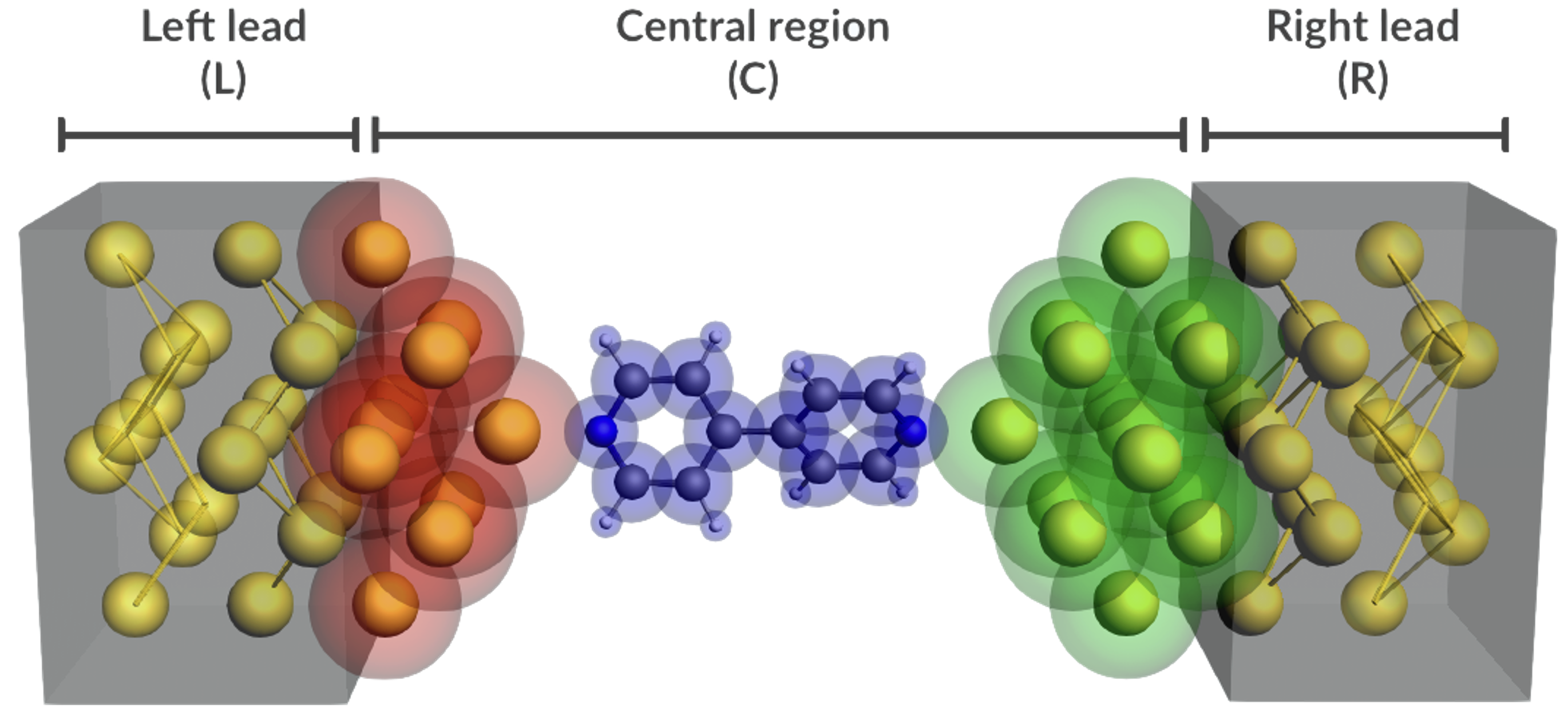
外場の設定: 一様な静電場(1〜2次元)、非平衡グリーン関数(NEGF)法
構造予測・反応解析
構造最適化計算: quasi-Newton
遷移状態探索: EF
プロパティ計算
バンド構造
状態密度: 全状態密度、部分状態密度、局所状態密度
原子電荷: Mulliken, Hirshfeld, Voronoi
数値微分による振動数計算
フォノン分散、熱力学物性(比熱、自由エネルギー)
周波数依存誘電関数: バンド間遷移とバンド内遷移の両方に対応
電子エネルギー損失分光スペクトル(EELS)
NMR: 化学テンソル
ESR: gテンソル、Aテンソル、NQCC(EFG)、Qテンソル
輸送特性: バンド伝導(有効質量に基づく輸送特性の評価)、バリスティック伝導(NEGF法によるI–V曲線の計算)
解析機能
結合エネルギー解析
電荷密度フーリエ解析
AIM, ELF
精度・効率
数値型+Slater型の原子基底関数: 周期表の全元素に対応した全電子基底関数およびFrozen Core基底関数を完備
電子密度フィッティングによるクーロン相互作用の高速化
BAND-GUI
BAND-GUIは、BAND用のグラフィカルユーザインターフェースです。
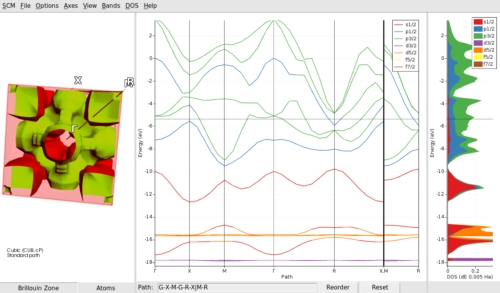
構造構築機能
ブラベー格子の指定による結晶構造の構築
ミラー面の指定による表面構造の構築
表示機能
バンド構造図の表示、ブリルアンゾーンの3次元表示、フォノン分散図の表示
3次元データの等値面・等高線表示: ブロッホ軌道、差電子密度など
アニメーション表示: 構造最適化計算、基準振動解析
計算設定・ジョブ管理機能
計算方法、基底関数、プロパティなどの計算設定
計算ジョブの投入・管理機能
COSMO-RS
COSMO-RSプログラムは、COSMO-RS (COnductor like Screening MOdel for Realistic Solvents) 法(*1)を搭載したADFのポスト計算プログラム(*2)です。
COSMO-RSプログラムには計算の設定および結果の可視化を行うためのグラフィカルユーザインターフェース(COSMO-RS-GUI)が付属し、ADFの計算結果をシームレスに利用することができます。
COSMO-RSプログラムは、ADFのCOSMO計算から得られた表面電荷密度を使用します。
COSMO-RSプログラムでは、任意の化合物の純物質および混合物に対して、以下の熱力学物性が計算できます。
活量係数
蒸気圧、沸点
分配係数
気液・液液平衡
溶解度
ヘンリー定数
過剰エネルギー
1) A. Klamt, V. Jonas, T. Burger, and J. C. W. Lohrenz, J. Phys. Chem. A, 102, 5074 (1998).
2) C.C. Pye, T. Ziegler, E. van Lenthe, and J.N. Louwen, Can. J. Chem. 87, 1 (2009).

COSMO-RS-GUI
COSMO-RS-GUIは、COSMO-RSプログラムのグラフィカルユーザインターフェースです。計算の設定から、結果の可視化までを行うことができます。
ReaxFF
反応分子動力学計算プログラム
ReaxFF は、ペンシルベニア州立大学のvan Duin 教授らによって開発された反応分子動力学計算プログラムです。ReaxFF は、既存の古典分子動力学プログラムとは異なり、結合の生成と開裂を記述することができる反応力場 (Reactive Force Field)(*1) を搭載しています。反応力場のパラメータは量子化学計算のデータから決められているために化学反応の高精度な記述を可能にする一方で、その動力学計算は通常の古典分子動力学シミュレーションに匹敵する計算速度を保持しており、触媒反応や燃焼反応など、材料科学分野における分子動力学シミュレーションの適用範囲を大幅に広げています。
SCM 社ではReaxFF コードに対して並列化を含む各種最適化を行っています。また、メモリ使用量などの最適化により10万原子程度を含む3 次元周期系の計算が通常のデスクトップPC上で対応可能です。また、ReaxFF にはグラフィカルユーザインターフェース(GUI)が用意されており、バルクモデルの構築、分子動力学の計算設定、ジョブ投入、計算結果の可視化に対応しています。
1) A.C.T. van Duin, S. Dasgupta, F. Lorant, and W.A. Goddard III, J. Phys. Chem. A 105, 9396 (2001).
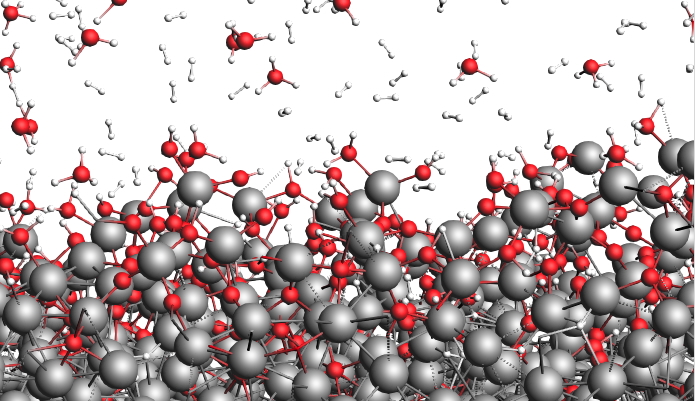
ReaxFFの主な機能
反応力場による構造最適化と分子動力学計算 (アンサンブルはNVE, NVT, NPT に対応)
3 次元バルクモデルの構築(Packmol を使用した溶媒分子のランダム配置に対応)
シミュレーティッドアニーリングおよび異なる温度領域の設定
分子動力学計算で得られた座標履歴のアニメーション表示と各種物理量の時間発展のグラフ表示
PyMDとのインターフェース(メタダイナミスクやアンブレラサンプリングなどの自由エネルギー計算に対応)
DFTB
密度汎関数法に基づくタイトバインディング計算プログラム
DFTB(Density Functional based Tight-Binding)は、密度汎関数法に基づくタイトバインディング計算プログラムです。DFTBでは、電子間相互作用の積分計算をパラメータ化することで高速かつ精度の高い計算が実現されており、通常の密度汎関数法計算では取り扱えない大規模な系にも適用することが可能です。DFTBパラメータは、Quasinano*プロジェクトによって開発されたパラメータを利用可能で、周期律表のほとんどの元素をサポートしています。DFTBのハミルトニアンでは、荷電系の正確な取り扱いを可能にするためのSelf-consistent charge (SCC-DFTB)法の搭載や、長距離相互作用を記述するための分散力補正などがなされています。
DFTBを利用することで、大規模な系に対しても長時間のシミュレーションを実行することがパソコンレベルの計算機で実現できます。DFTBは周期系だけでなく孤立分子系にも対応しています。より高精度な計算(密度汎関数法など)を実行する前の簡易構造最適化計算プログラムとしてDFTBを利用することもできます。SCM社製ソフトウェアでは密度汎関数プログラムとしてADFやBANDが利用可能で、これらのプログラムと連携した取り扱いが可能です。
*) Quasinanoプロジェクトは、独Jacobs大学と蘭SCM社の共同研究による取り組みで、軽元素だけでなく遷移金属も含むナノサイズの系(10万原子程度)を高速かつ精確に扱える手法の開発を目指したプロジェクトです。
DFTBの主な機能
計算方法
・Self-consistent charge (SCC-DFTB)法
・third-order self-consistent charge (DFTB3) 法: DFTB.orgパラメータのみ対応
・分散力補正(D3, D3-BJ, UFF)
・時間依存(TD-DFTB)法
・非平衡グリーン関数(NEGF-DFTB)法
DFTBパラメータ
・QUASINANO (周期律表の多くの元素に対応:下図参照)
・Dresden set (Al-O-P-C-H, Al-Si-O-H 等)
・DFTB.org (H-C-N-O-S-P, Si-F-O-N-C-H 等)
・GFN1-xTB (周期律表の多くの元素に対応)
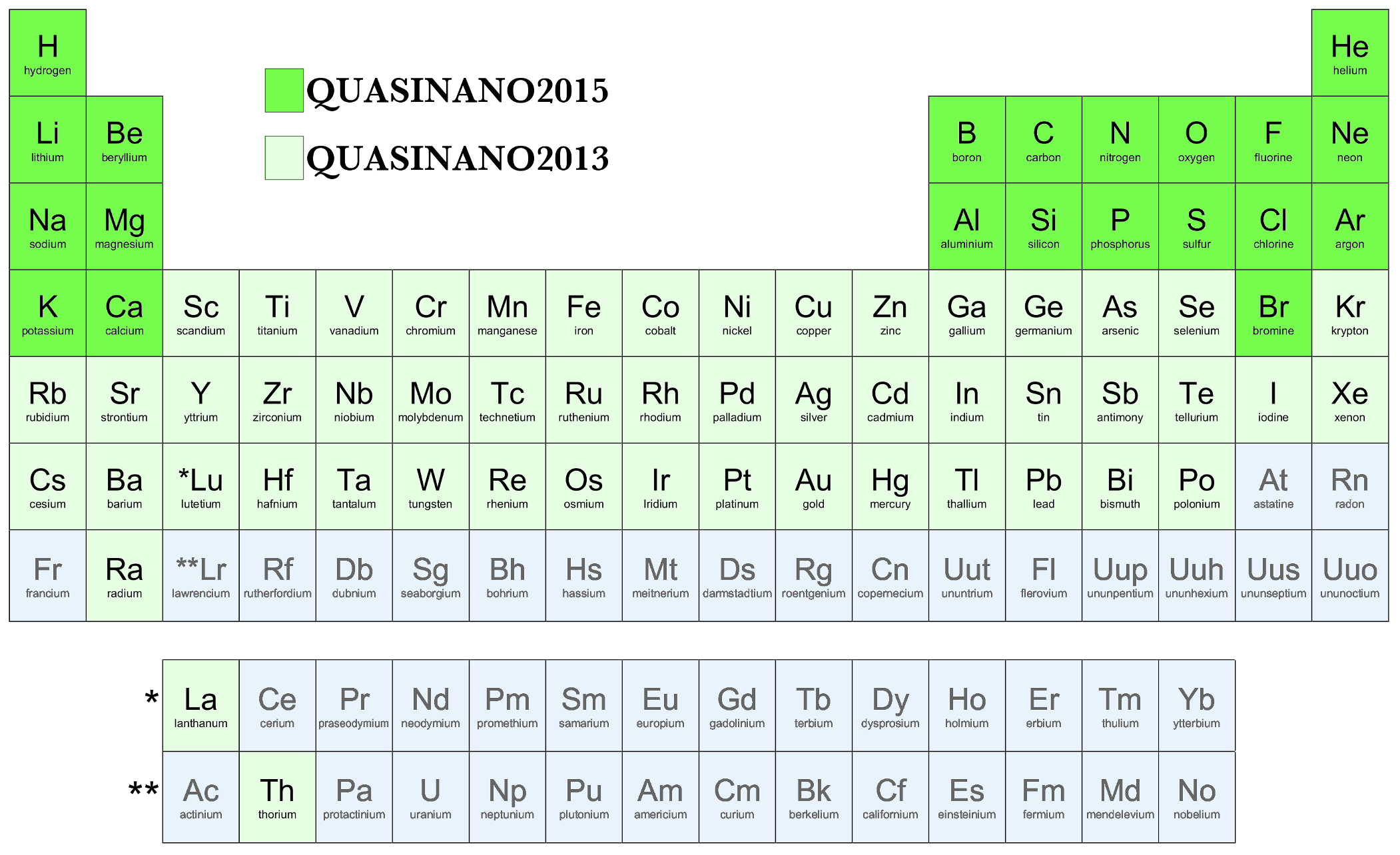
QUASINAN02013: 電子ハミルトニアンのパラメータ。バンド構造、DOS、UV/VIS、NEGFなどの電子特性の一点計算をサポートします。 [M. Wahiduzzaman et al. J. Chem. Theory. Comput. 9, 4006 (2013)]
QUASINAN02015: 反発パラメータを追加。構造最適化、IRスペクトル、MDなどの計算をサポートします。 [A. F. Oliveira et al. J. Chem. Theory. Comput. 11, 5209 (2015)]
MLPot (ML Potentials)
機械学習ポテンシャルに基づくMD & MC 計算エンジン
MLPotは、機械学習ポテンシャル(MLP)技術を利用して、原子・分子系のエネルギー評価や動力学シミュレーション(分子動力学法:MD、モンテカルロ法:MC)を実行するためのソフトウェアです。これは、ニューラルネットワークなどの機械学習モデルを用いて、原子配置から系のポテンシャルエネルギーを予測する手法です。MLPのアプローチにより、量子化学計算に近い精度を維持しながら、古典的な力場計算のような高い計算効率でのシミュレーションの実行可能性を高めます。
標準搭載されている基盤モデル
MLPotには、様々な研究対象に適用できるよう、事前に訓練された複数の「基盤モデル」が標準でパッケージ化されています。以下は、搭載されている主なモデルとその特徴です。
• ANI モデル (ANI-1x, ANI-1ccx, ANI-2x)
主に有機分子のエネルギーや構造解析に用いられます。対象元素はH, C, N, O, F, Cl, Sなどで、複数のモデル予測を統合する「委員会モデル」を採用しており、予測の不確かさを評価できる点が特徴です。
• AIMNet2 モデル
有機・無機分子およびイオンを対象とし、HからIまでの主要な元素(H, B, C, N, O, F, Si, P, S, Cl, As, Se, Br, I)に対応します。分散相互作用や静電相互作用を考慮したモデルです。
• M3GNet モデル
固体や結晶材料の研究に適しており、周期表の広範な元素をカバーします。Materials Projectのデータを基に訓練されているため、多様な無機材料系の計算に利用できます。
これらの基盤モデルは、GPUアクセラレーションに対応しています。
外部モデルとの連携機能
MLPotは、外部で開発された、あるいはユーザー自身が構築したMLPモデルを柔軟に組み込むためのインターフェースを備えています。この連携は、計算化学分野の標準的なプラットフォームである ASE (Atomic Simulation Environment) を介して行われます。ASEに対応したインターフェースを持つ外部のMLPモデルやパッケージであれば、MLPotのシミュレーションエンジンと結合して使用することが可能です。
• 連携が想定される公開モデル
CHGNet, FairChem, MACE, MatterSim, ORBなど
• 連携が想定されるパッケージ
NequIP, PiNN, SchNetPack, SGDML, DeePMD-kit など
この拡張性により、ユーザーは特定の研究目的に特化した最新のモデルを自身のシミュレーションワークフローに取り入れることができます。
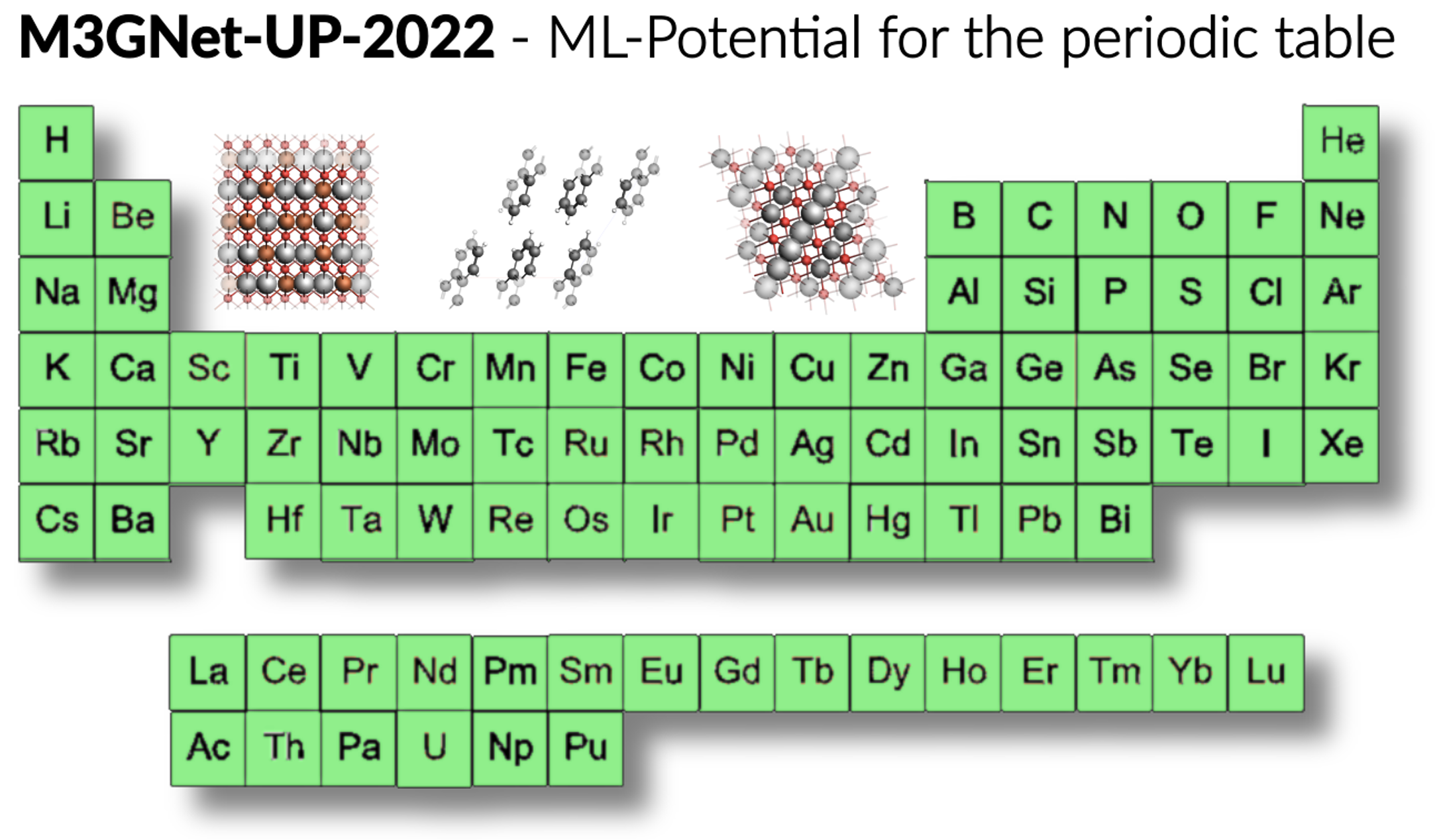
FF (ForceFields)
古典的力場計算エンジン
FFは、分子系のシミュレーションを近似的でありながら高速に行うための、古典的な力場を集めたソフトウェアです。原子の電荷を固定して扱うモデルから、周囲の環境に応じて電荷の偏りを考慮する分極可能なモデルまで、複数の選択肢を提供します。
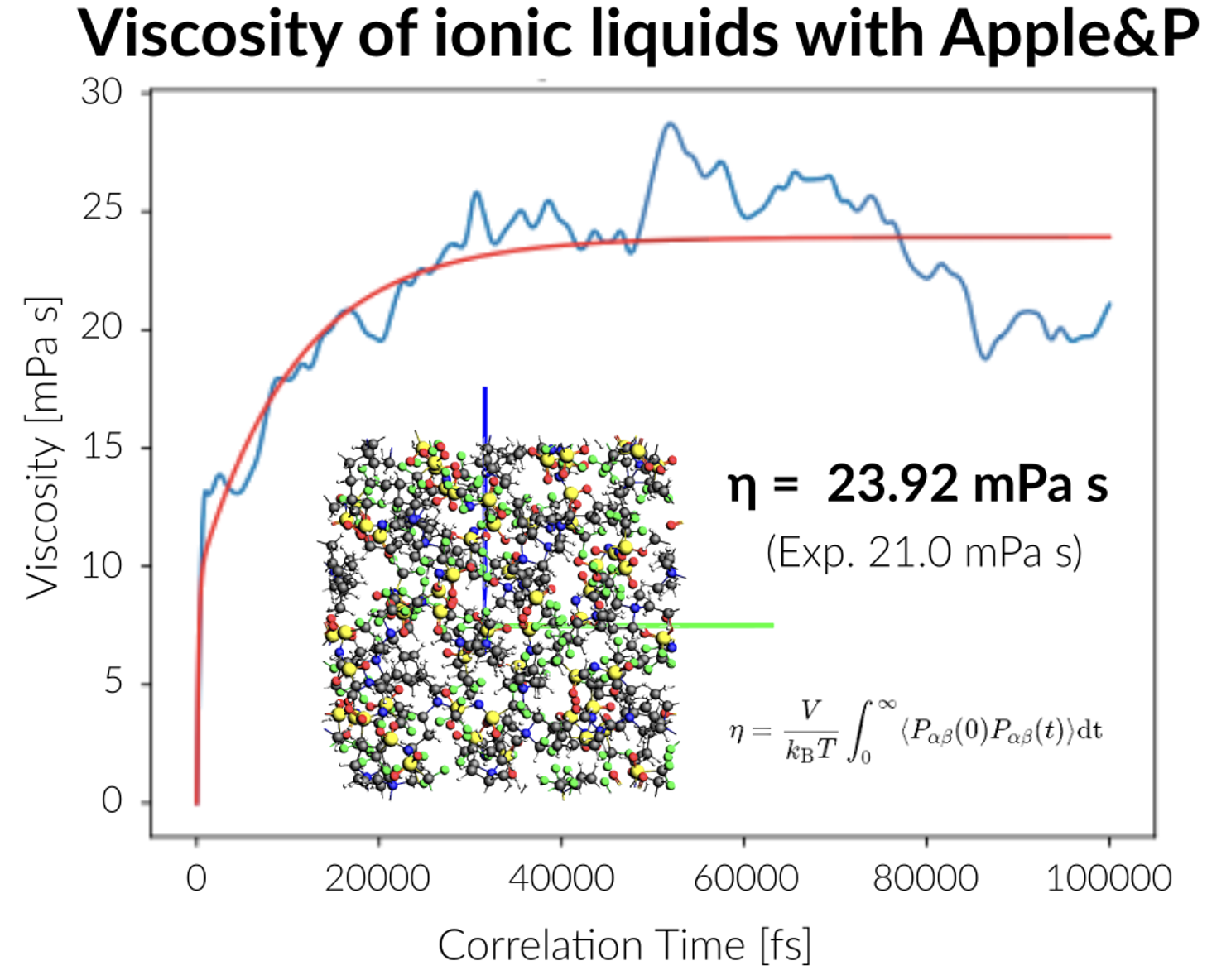
分極性力場の一つとして、SpicherとGrimmeによって開発されたGFN-FFを搭載しています。これは周期表のほとんどの元素に対応可能な自動化された力場であり、量子化学計算に近い精度と計算速度の両立を目指して設計されています。同じく分極性力場であるAPPLE&Pは、特にイオン液体や電解質、多くのポリマー系のシミュレーションに適したパラメータを備えています。
※APPLE&Pのパラメータは別途ライセンスの取得が必要です。
これらの他に、分極性でない力場として、汎用的に利用できるUFF、生体分子で実績のあるAmber95、材料分野向けのTripos 5.2、有機小分子に適したGAFFなども利用可能です。
FFを用いることで、高度な分子動力学(MD)およびモンテカルロ(MC)シミュレーションを実行できます。また、AMSのハイブリッドエンジンを介して、これらの力場をQM/MM計算の一部として利用することも可能です。
FFの計算エンジンはGUI(グラフィカルユーザーインターフェース)によって完全にサポートされており、シミュレーションの設定、実行、結果の解析を直感的に行えます。物性予測や計算軌道の解析といった機能も、追加の設定なしですぐに利用を開始できます。ユーザー向けには、チュートリアル、具体的なワークフロー、解説ビデオなど、豊富なドキュメンテーションが用意されています。
title:{コア機能}
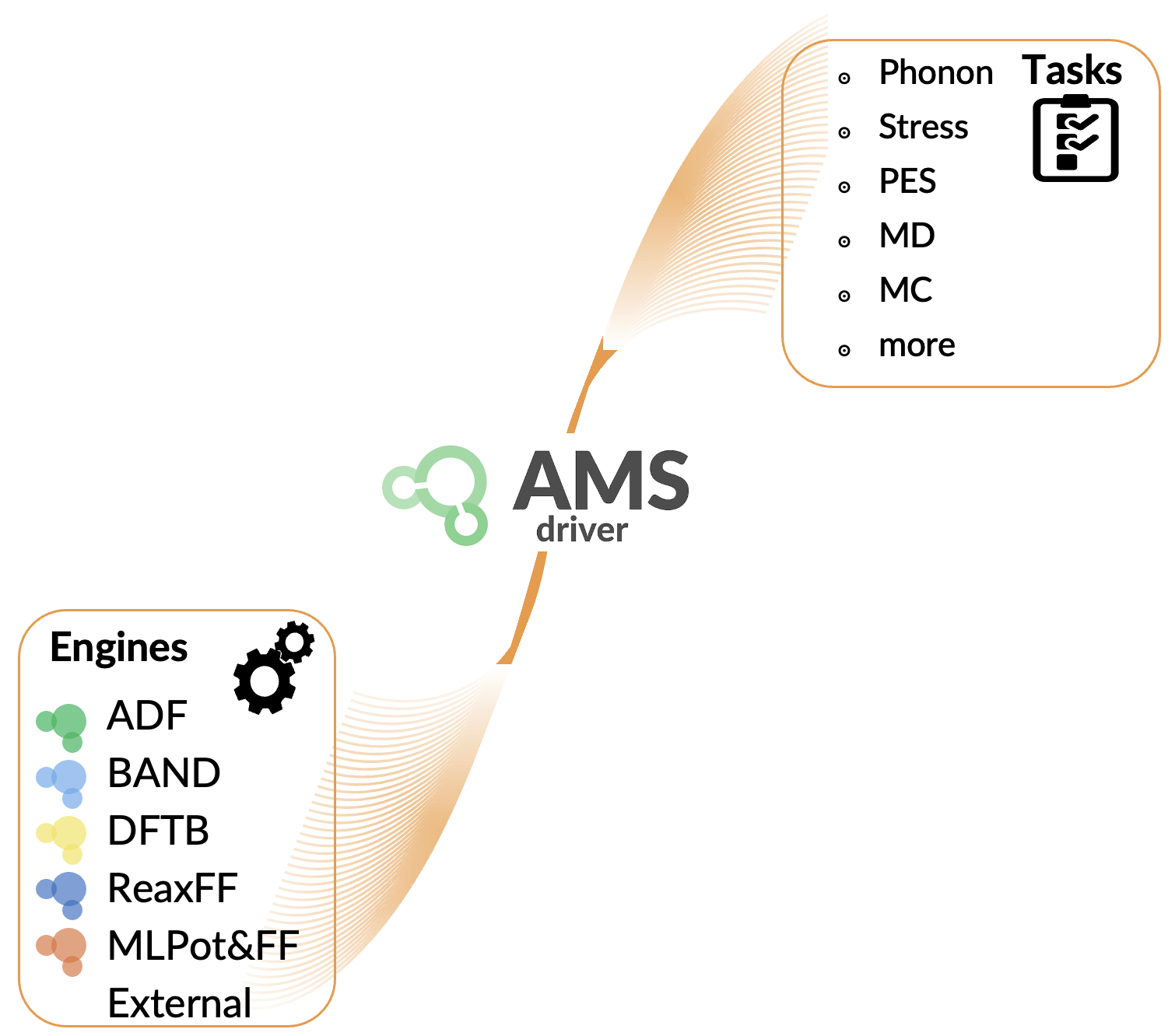
AMSドライバー
AMSドライバーは、分子シミュレーションにおける高レベルなタスクを管理・実行する、AMSの中核をなすモジュールです。その最も基本的な入力は研究対象となる化学系の構造情報であり、その役割は系のポテンシャルエネルギー曲面(PES)を記述し、探索することにあります。ドライバーは、ユーザーが指定した計算エンジン(ADF、DFTB、古典力場など)を用いてエネルギーや力を算出し、それに基づいて構造最適化や分子動力学(MD)計算といったタスクの中で、系の構造変化を体系的に処理します。
ドライバーとエンジンの連携プロセス
典型的なワークフローは以下の通りです。
1. ユーザーは実行したいタスク(例:構造最適化)と使用するエンジン(例:DFTB)を指定します。
2. AMSドライバーは現在の構造をエンジンに渡します。
3. エンジンはエネルギーや原子に働く力の勾配などを計算し、その結果をドライバーに返します。
4. ドライバーは結果を解析し、タスクの目的に応じて計算を終了するか、あるいは構造を更新してステップ2に戻り、計算を続行します。
このアーキテクチャは非常に柔軟であり、AMS内部のエンジンだけでなく、外部プログラムをエンジンとして利用することも可能です。
計算出可能な物性(プロパティ)
AMSドライバーは、計算エンジンが提供するエネルギー、力、応力テンソルといった基本的な物理量を用いて、以下のような多様な物性を算出します。
・熱力学的特性: エンタルピー、エントロピー、自由エネルギーなど
・弾性: 弾性テンソルや各種弾性率
・振動スペクトル: IR(赤外吸収)、ラマン
・フォノン: 結晶における格子振動とそれに関連する物性
また、外部の計算コードや自作のプログラムをエンジンとして接続するためのインターフェース(ASE準拠)も備えており、高い拡張性を持ちます。
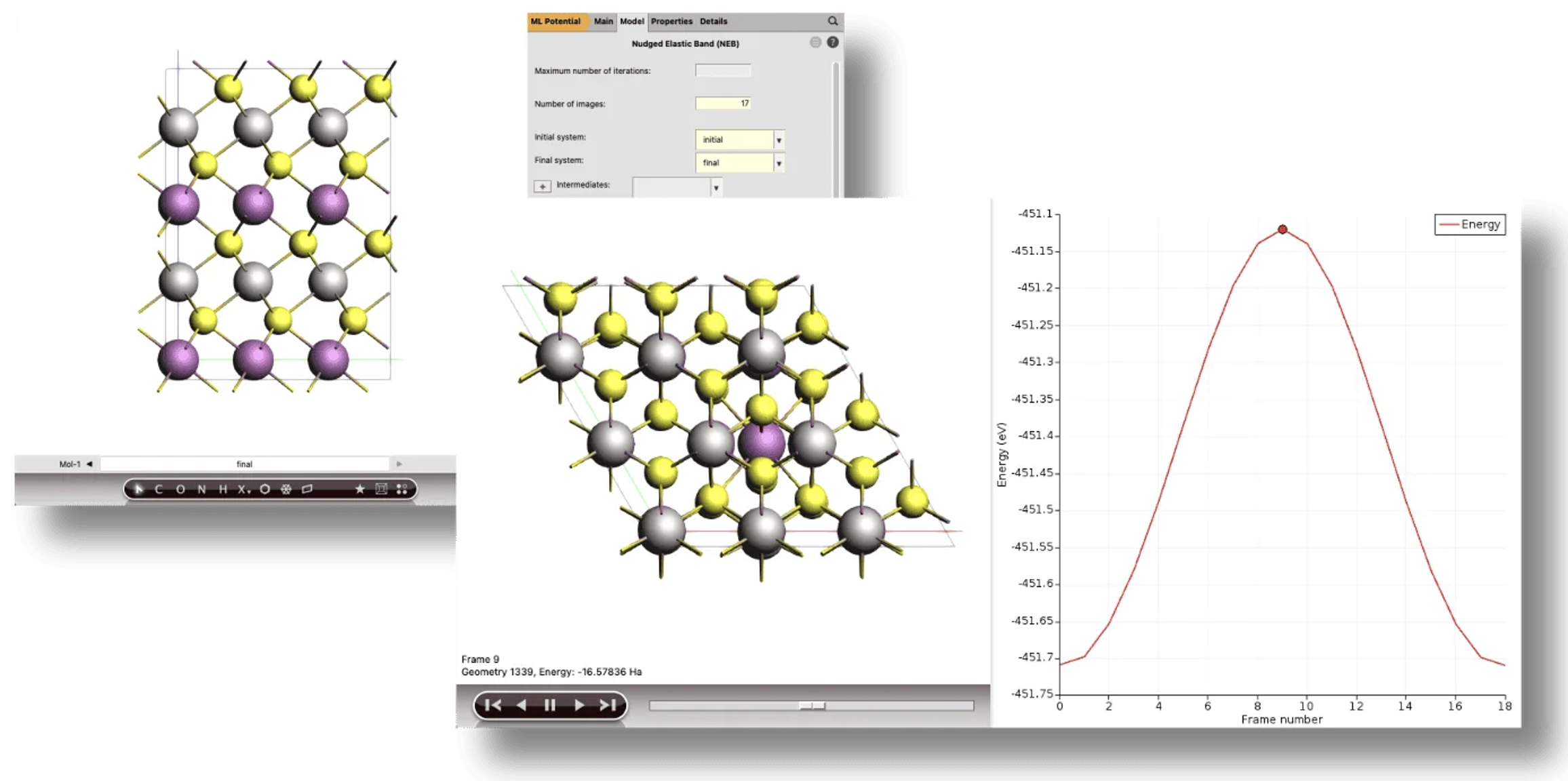
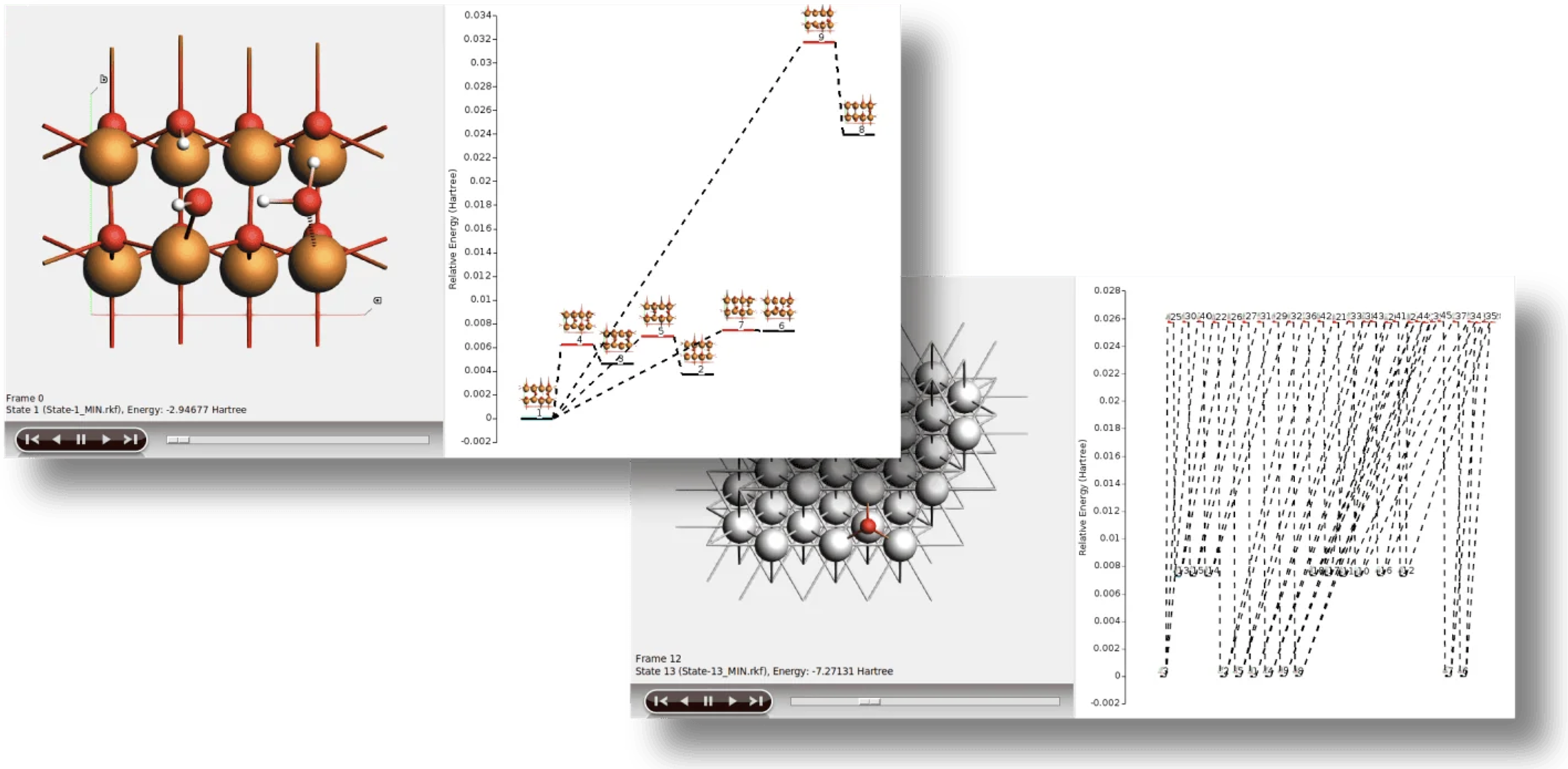
ポテンシャルエネルギー曲面の探索(PES探索)
系の安定構造(局所的極小点)や化学反応の遷移状態(鞍点)など、PES上の特異点を自動的に発見するためのアルゴリズム群です。
・プロセス探索(Process Search): ある安定状態からの脱出経路(メカニズム)を、極小点と鞍点の両方を含めて発見する複合的な手法
・ベイシンホッピング(Basin Hopping): モンテカルロ法に基づき、効率的に多数の局所的極小点を見つけ出し、大域的最小構造を探索する手法
・鞍点探索(Saddle Search): ある構造の近傍に存在する鞍点(遷移状態)を見つけ出すための片側探索法
・ランドスケープ精密化(Landscape Refinement): 事前に計算されたエネルギー地形に対し、より高精度な計算設定で各構造を再最適化する機能
・結合サイト(Binding Sites): 事前に計算されたエネルギーランドスケープの情報から、分子間の結合サイトを特定し、計算する機能
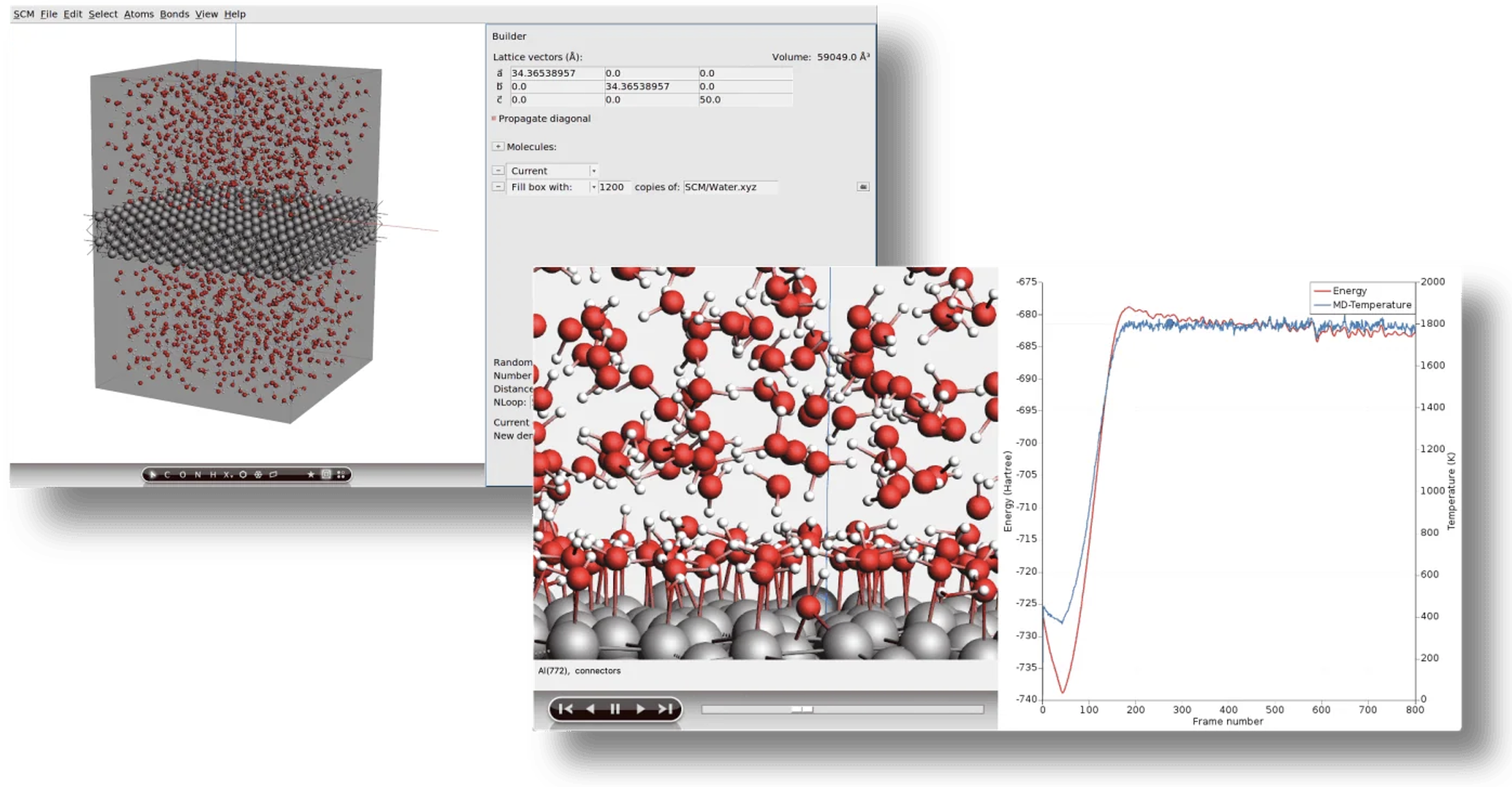
分子動力学(Molecular Dynamics, MD)
ニュートンの運動方程式を数値的に解くことで、系の時間発展を追跡するシミュレーション手法です。AMSは多彩なMD機能と高い並列計算性能を誇ります。
・GUIによる複雑な系の簡単なセットアップ: 溶媒和、ポリマー、結晶構造などの複雑な系を、グラフィカルなインターフェース上で直感的に構築
・様々な温度・圧力制御(サーモスタット・バロスタット): シミュレーション中の温度や圧力を一定に保つための、多様な制御手法
・MDの加速と長時間シミュレーション: 反応座標駆動ハイパーダイナミクス(CVHD)やレプリカ交換分子動力学(REMD)など、長時間スケールの現象を効率的にシミュレーションするための各種手法
・PLUMEDを介したメタダイナミクス: 外部ツールPLUMEDと連携し、自由エネルギー地形を効率的に探索するメタダイナミクスを実行
・Bond Boost法によるポリマーの架橋反応促進: Bond Boost法を用いることで、ポリマー鎖間の架橋反応シミュレーションを加速
・分子ガンと分子シンク: CVDやスパッタリングといったプロセスを模し、指定した領域に原子や分子を射出・吸収させる機能
・非平衡分子動力学(NE-MD): 熱伝導(T-NEMD)など、平衡状態にない系のダイナミクスをシミュレーション
・トラジェクトリ解析ツール: 動径分布関数(RDF)、拡散係数など、シミュレーション結果を分析するための豊富なツール群
・強力なスクリプトインターフェース: 一連の計算ワークフローを自動化するための、柔軟で強力なスクリプト機能
・高性能なハイブリッド並列計算: MPIとOpenMPを組み合わせたハイブリッド並列化により、計算リソースを最大限に活用
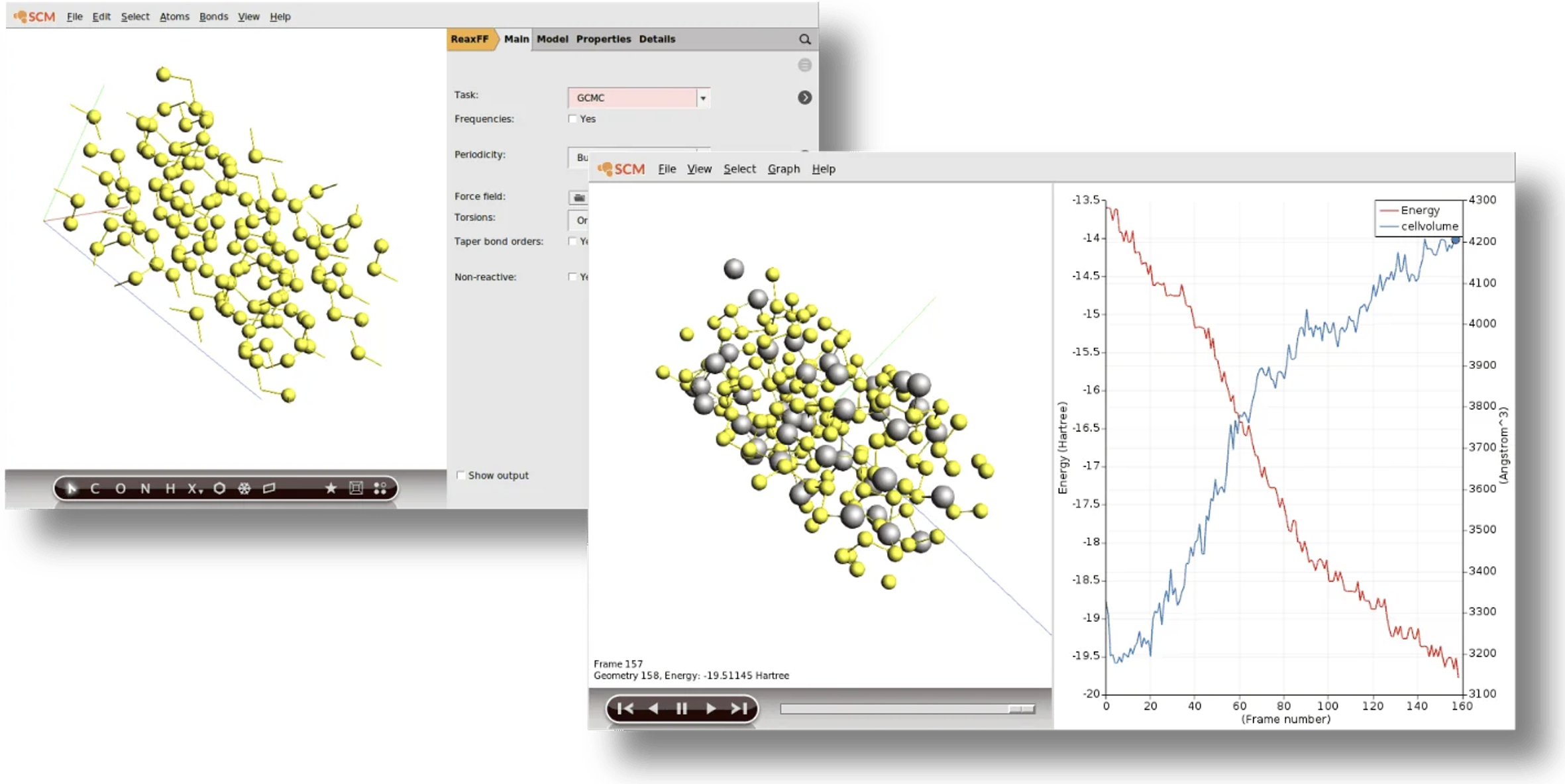
モンテカルロ法(GCMC, fbMC)
確率論的な手法を用いて、系の統計的な振る舞いを調べるシミュレーションです。
グランドカノニカル・モンテカルロ法(GCMC)
外部と粒子のやり取りがある系(開いた系)を扱います。電池の充放電プロセスや、多孔質材料へのガス吸着等温線の計算などに利用されます。
フォースバイアス・モンテカルロ法(fbMC)
原子に働く力を利用してシミュレーションを効率化する加速手法です。原子層堆積(ALD)、表面拡散、薄膜成長、結晶の欠陥修復(ヒーリング)過程など、速度の遅い現象の研究に用いられます。
title:{ワークフロー}
Advanced Workflowsについて
Advanced Workflows(先進的ワークフロー)は、計算化学統合パッケージAMS(Amsterdam Modeling Suite)に搭載された、一連の高度な計算タスクを自動化・効率化するためのツール群です。複数の計算ステップをシームレスに連携させることで、複雑な物理現象や化学現象のシミュレーションを可能にし、研究開発のプロセスを大幅に加速させます。
主なワークフローツール
Advanced Workflowsには、特定の研究目的に特化した以下のツールが含まれています。
・ParAMS: 各種計算モデルのパラメータ最適化を支援します。
・ChemTraYzer2: 分子動力学計算の軌跡(トラジェクトリ)を解析し、反応ネットワークを抽出します。
・OLED workflows: 有機EL(OLED)材料の性能評価を多角的に行います。
・ACE-Reaction: 反応物から生成物への可能な反応経路を、結合の切断と形成に基づいて網羅的に探索するツールです。
・AMSkinetics (MKMCXX / Zacros):
マイクロキネティクスや速度論的モンテカルロ法を用いて、触媒反応などの速度論を解析します。
ParAMS (Parameters for AMS)
高精度なモデル開発を支えるパラメータ最適化ツール
ParAMS(パラムスと読みます)は、原子や分子のシミュレーションで使われる様々な計算モデル(エンジン)のパラメータを最適化するための柔軟な支援ツールです。使いやすいグラフィカルユーザーインターフェース(GUI)とPythonライブラリを提供し、パラメータ化のプロジェクトをスムーズに進められるように支援します。
特に、以下のモデルのパラメータ化に特化したインターフェースを備えています。
・ReaxFF:
Reactive Force Field / 反応性力場
・DFTB
Density-Functional Tight-Binding / 密度汎関数強束縛法
・MLPot
Machine Learning Potentials / 機械学習ポテンシャル

このツールの目的は、研究対象の化学システムに対してモデルの予測精度を上げるために、既存のパラメータを再調整(リフィット)したり、新しいパラメータを開発したりすることです。

ParAMS GUIの設定・表示例
ParAMSの主な機能
ParAMSは、高精度なモデル開発のプロセスを、以下の各段階で支援する機能を提供します。
1. 訓練セットの構築
パラメータ最適化の基礎となるのは、モデルが学習するための「正解データ」を含む訓練セット(Training Set)です。ParAMSは、この訓練セットを準備する上で高い柔軟性を持つように設計されています。
参照データとして、実験値や、より高精度な参照計算(例:密度汎関数理論計算)から得られる物理量を用いることができます。例えば、一点計算から得られるエネルギーや原子に働く力、構造最適化後の最適な構造(結合長・角度など)、あるいは化学反応経路に沿った相対的なエネルギー変化など、多様なデータを訓練データとして利用可能です。
また、モデルが訓練データに適合しすぎる「過学習(Overfitting)」という現象が起きていないかを確認するため、訓練セットとは別の検証セット(Validation Set)を使うことができます。検証セットに対する誤差は直接最適化されませんが、その値を監視することで、モデルが未知のデータに対しても高い予測性能を持つか(汎化性能)を評価する指標となります。
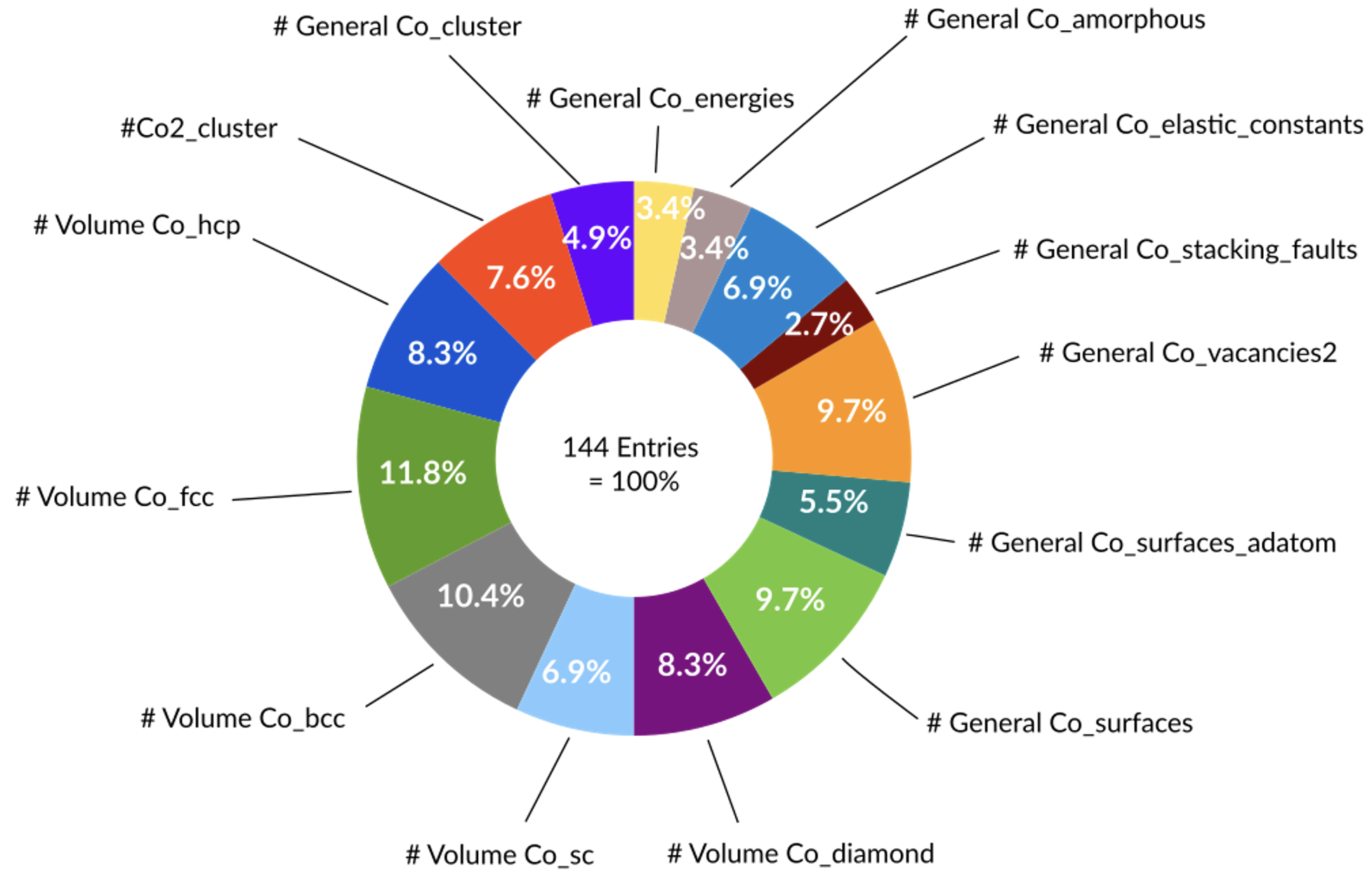
Co(コバルト)の訓練セット
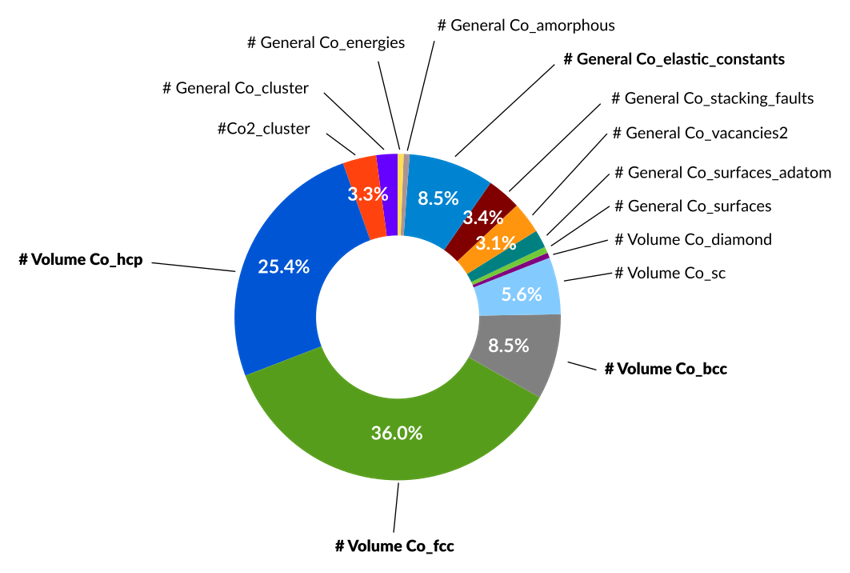
Co 訓練セット(重み付き)
2. パラメータ最適化と感度分析
訓練セットの準備ができたら、モデルのパラメータを最適化します。
最適化アルゴリズム:
ReaxFFとDFTBの最適化は、GloMPO(Globally Managed Parallel Optimization)という仕組みを通じて実行されます。これは、多数の最適化計算を並行して実行し、結果の思わしくない計算を自動で停止・再起動することで、効率的に最適解の探索を管理する手法です。標準のアルゴリズムとしてCMA-ES(Covariance Matrix Adaptation Evolution Strategy; 共分散行列適応進化戦略)が採用されています。これは進化的アルゴリズムの一種で、目的関数の数学的な傾き(勾配)を必要としないため、複雑でノイズの多いパラメータ空間の探索にも適しています。
損失関数(Loss Function):
最適化は、「損失関数」と呼ばれる指標の数値を最小にすることを目指します。損失関数は通常、訓練データに含まれる「正解(参照)値」と「モデルによる計算値」との間の誤差(重み付き二乗誤差)の合計として定義されます。このとき使われる重みパラメータ σ は、エネルギーや力といった単位の異なる物理量の誤差を公平に扱えるように、それぞれの重要度を調整する役割を持ちます。
感度分析(Sensitivity Analysis):
特にReaxFFのように多くのパラメータを持つモデルでは、どのパラメータが結果に大きく影響するかを特定することが重要です。ParAMSは、HSIC(Hilbert-Schmidt Independence Criterion)という手法に基づくグローバル感度分析ツールを実装しています。これは、モデルの性能に対して最も影響の大きいパラメータ群を統計的に特定する手法です。感度の高い重要なパラメータだけに絞って最適化を行うことで、計算時間を短縮し、より安定した最適化と過学習の抑制が期待できます。
3. アクティブラーニングによる訓練セットの拡張
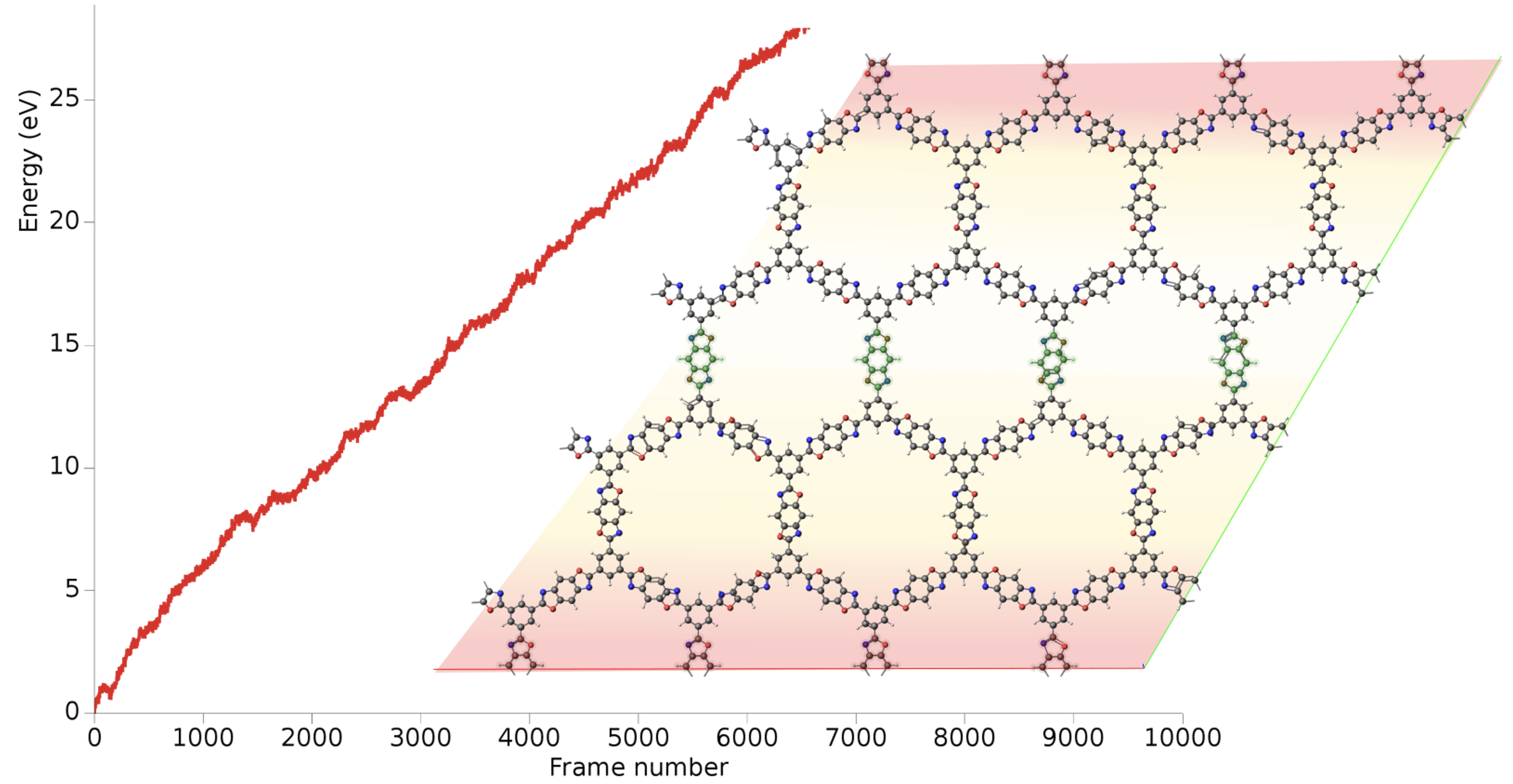
機械学習ポテンシャルの精度は、学習に用いる訓練データの質と量に大きく左右されます。精度の高い参照データの計算には多大なコストがかかるため、効率的なデータ収集が不可欠です。そこでParAMSは、アクティブラーニング(Active Learning; 能動学習)のワークフローをサポートしています。
アクティブラーニングは、モデル自身が「次にどのデータを学習すれば効率的に賢くなれるか」を判断し、データを要求することで、訓練セットを反復的に賢く拡張していく手法です。
このワークフローでは、まず今あるデータでモデルを構築し、そのモデルで分子動力学(MD)シミュレーションなどを行います。その中で、モデルが「この構造は学習したことがなく、予測に自信がない」と判断した場面(予測不確かさが高い領域)を特定します。不確かさの評価には、複数のモデルの予測のばらつきを利用する委員会モデル(Committee Model)などの手法が利用可能です。
そして、その「予測に自信がない」構造についてのみ高精度な参照計算を行い、新しい訓練データとして追加します。このサイクルを繰り返すことで、参照計算のコストを最小限に抑えながら、モデルの精度と適用範囲を体系的に向上させることができます。
アクティブラーニングによる最適化例
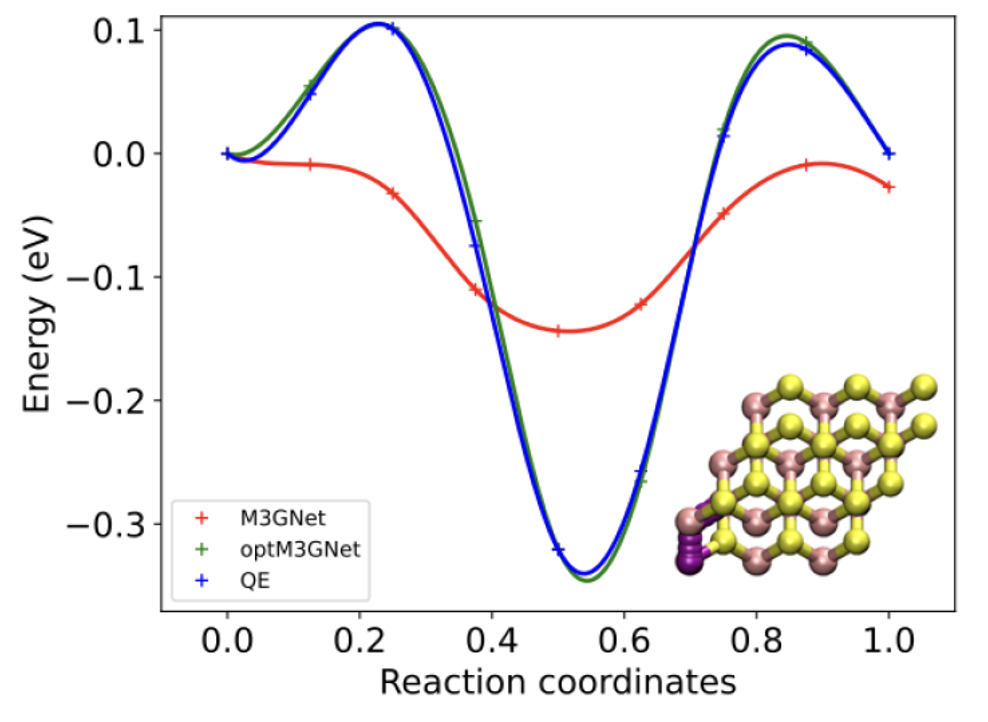
Li migration in MoS2
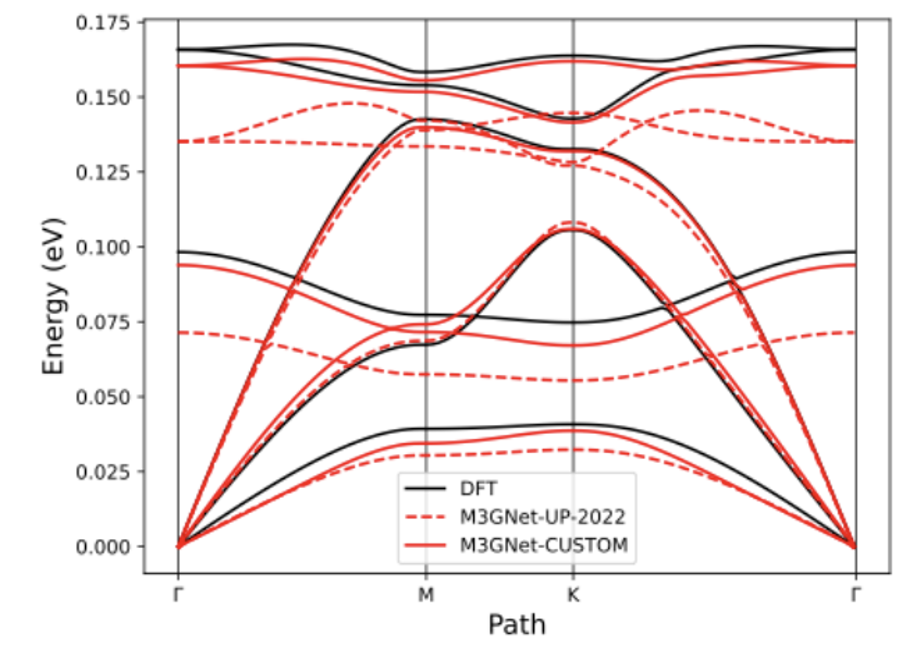
hBN phonons
ParAMSのライセンスについて
ParAMSは、「Advanced Workflows」のライセンスに含まれています。また、このツールでパラメータ化を行いたい計算エンジン(ReaxFF、DFTB、MLポテンシャル)のライセンスが別途必要です。
title:{Bumblebee}
Bumblebee
Bumblebeeは、OLEDスタック向けの3D-kMC(運動学的モンテカルロ)シミュレーションツールです。
Amsterdam Modeling Suite(AMS)とBumblebeeを組み合わせることで、有望なOLED材料およびデバイスのデジタルスクリーニングと予測を行うための、完全に統合されたマルチスケールシミュレーションプラットフォームが提供されます。
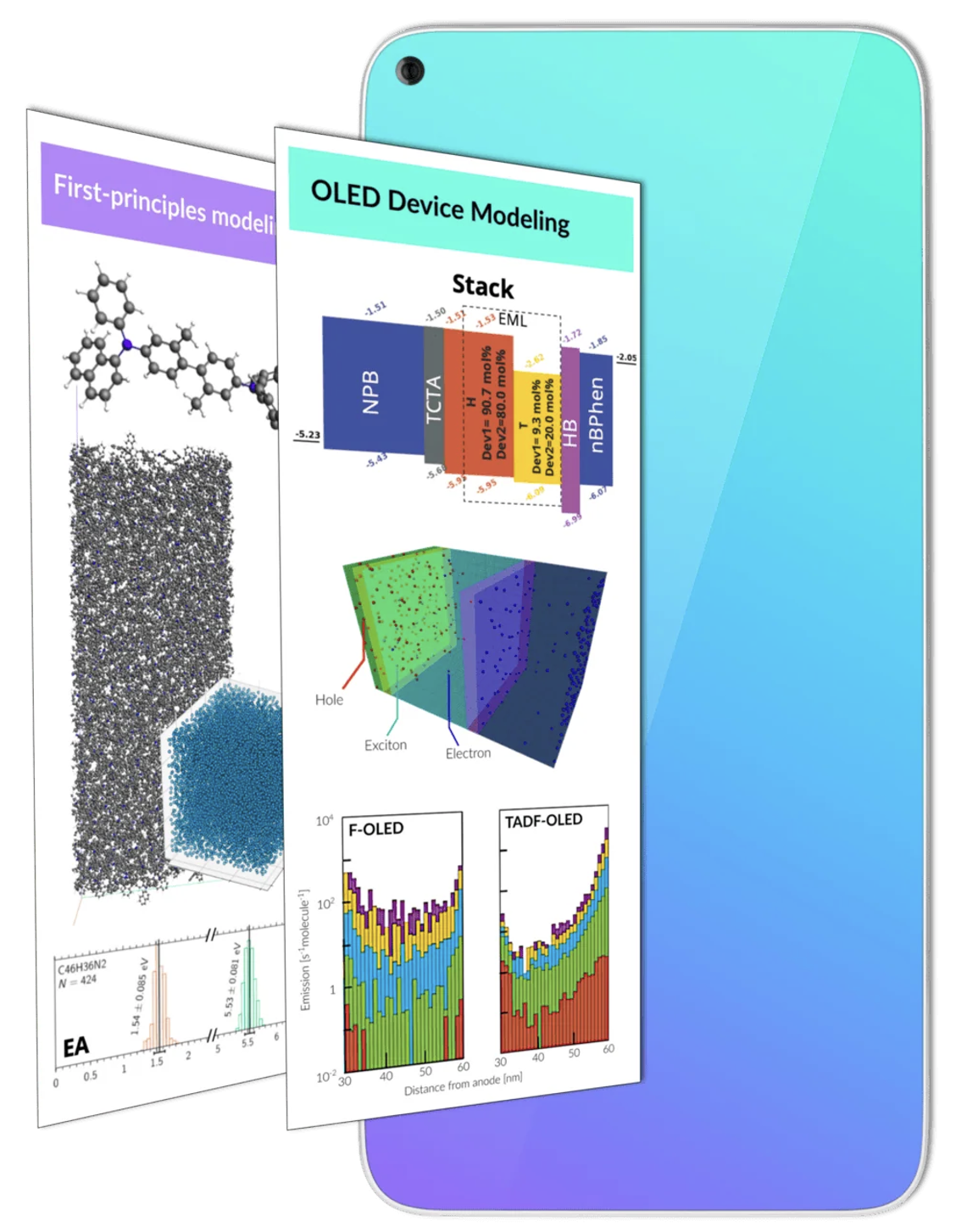
OLEDデバイスモデリング
Bumblebeeは、キャリアの拡散、分子発光、デバイス劣化を時間とともに追跡することで、OLED材料の長期的な挙動をモデル化するシミュレーションツールです。研究者は、このツールを用いて欠陥の形成プロセス、材料の経年変化、OLED層内での電荷キャリアの移動を理解し、デバイスの効率や寿命を向上させるための知見を得ることができます。
1次元ドリフト拡散モデルを超えたOLEDデバイスモデリング
• スタック内のナノ構造を考慮し、フィラメント輸送、バルクヘテロ接合、色素増感相互作用、高分子配向、ドーパントを含めたモデル化
• 非晶質の無秩序によるエネルギー状態密度(DOS)や遷移双極子配向のばらつきを考慮
• 分子間の静電相互作用を明示的にモデル化(長距離成分を含む)
• ホストとゲスト分子間の励起子の分布、移動、捕獲をモデル化
• 過渡応答シミュレーション、小信号解析、パルス実験をサポート
特徴
• TADF発光材料やハイパーフルオレッセンスなど、最先端のOLED材料をモデル化可能
• 明示的なモルフォロジーと時間変化の追跡により、スタック内の各プロセスの位置を特定
• ホスト・ゲスト分布、層界面、ドーパント勾配、高分子ネットワークなどの複雑な構造をモデル化
• 励起子消滅、ポーラロン消光、トリプレット収穫、エキシプレックス、振動結合などの複雑な光電子プロセスを考慮
• 分子の無秩序や長距離静電相互作用を含めたシミュレーションが可能
• 光取り出しシミュレーションにより、光管理の最適化を支援
• 新アルゴリズムにより、標準的な運動学的モンテカルロ法と比較して計算コストを10〜50倍削減
• パラメータスクリーニングや最適化ツールを内蔵し、スタック設計を最適化
OLEDのマルチスケールモデリング
AMSと組み合わせた第一原理OLEDデバイスシミュレーション
• 物理蒸着シミュレーションを用いて、現実的な薄膜構造を生成
• 薄膜内の全分子におけるイオン化ポテンシャル、電子親和力、双極子モーメントなどの分布を計算
• これらの特性をBumblebeeに入力し、OLEDデバイスシミュレーションを実行
適用事例
Multiscale modeling of OLED devices (SCM社ウェブサイト)
title:{適用事例}
適用事例
ランタノイド化合物・アクチノイド化合物の32電子則
遷移金属錯体は、18電子則を満たす化合物が安定であることが経験的に知られています。一方、fブロック元素(ランタノイド、アクチノイド)を含む化合物は、18電子則の類推から必然的に、32電子則を満たす化合物が安定であることが予想されます。最近、Dognonら(*1, *2)は、一連のアクチノイド化合物に対してADFを使用したDFT計算を行うことにより、32電子則を満たす化合物の安定性を確かめました。
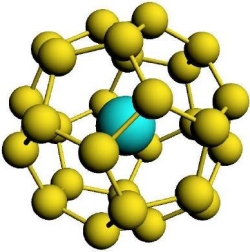
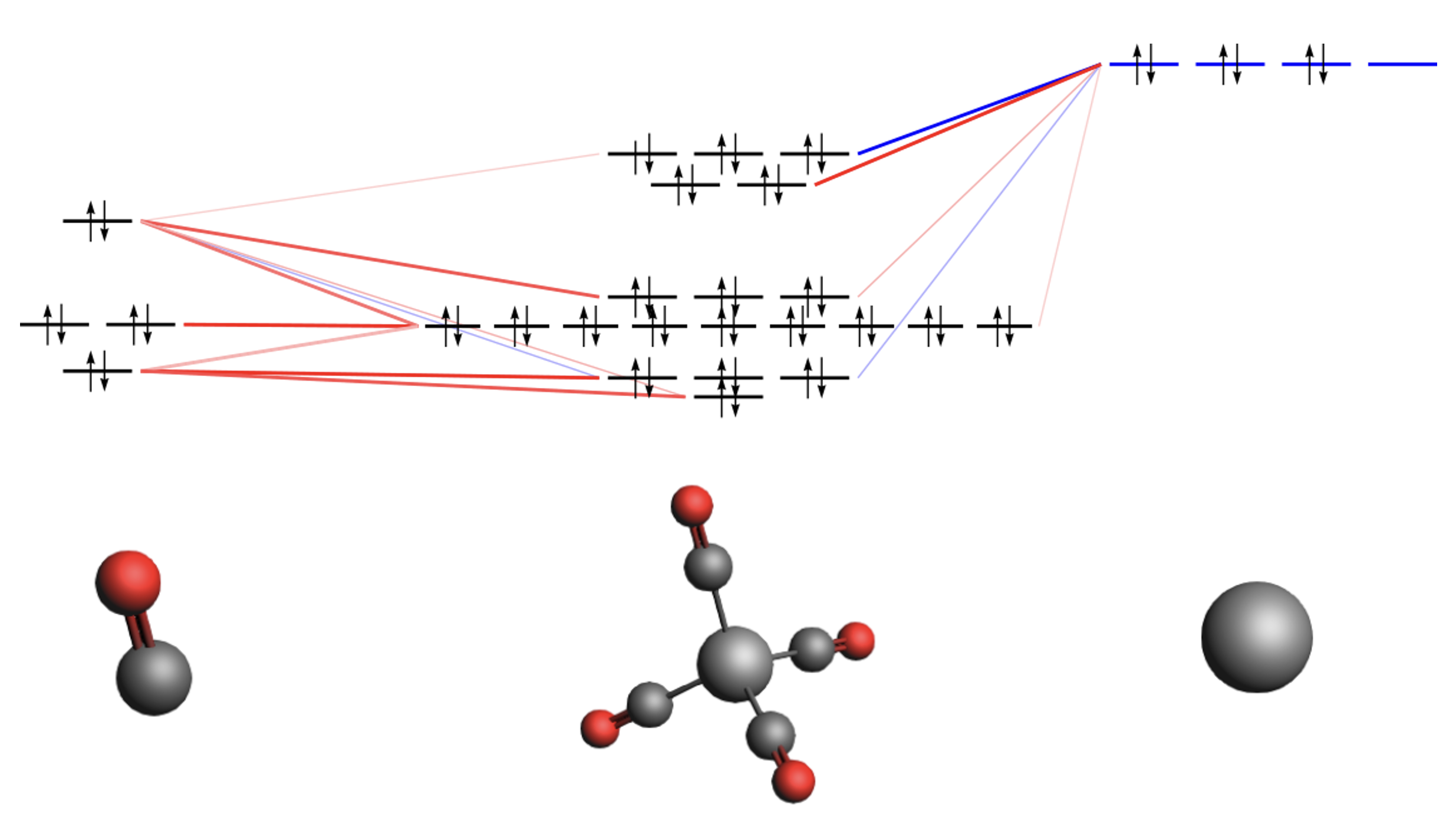
Pu4+@C28クラスター
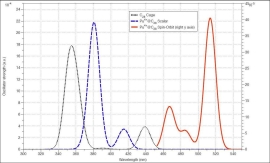
UV/Visスペクトル:スピン-軌道相互作用の効果を含む計算と含まない計算を比較
彼らは、エネルギー計算による分子の安定性評価の他に、IRやUV/Visなどのスペクトル計算も行っています。上図は、C28フラーレンに内包されたPu4+の系(左)とそのUV/Visスペクトル(右)を表示したものです。UV/Visスペクトルは、スピン-軌道相互作用を考慮した相対論とスカラー相対論の場合とで比較されていますが、このようなアクチノイド化合物に対してはスピン-軌道相互作用の効果が非常に重要であることがわかります。ADFでは、ZORA法と呼ばれる相対論法が実装されており、スピン-軌道相互作用を考慮した計算が可能です。
1) J. P. Dognon et al., Angew. Chem. Int. Ed., 46, 1427 (2007).
2) J. P. Dognon et al., J. Am. Chem. Soc., 131, 238 (2009).
鉄錯体のスピン状態間のエネルギー評価における基底関数の重要性
ADFの特長の一つとして、基底関数として一般的なGauss型ではなくSlater型の軌道を採用することで精度の高い計算を実現していることが挙げられます。 Güellら1は、ニトリルへドラターゼ(Nhase)のモデル錯体を始めとする一連の鉄錯体に対して、スピン状態間のエネルギー差の基底関数依存性をSlater型軌道(STO)とGauss型軌道(GTO)で比較しました。
GTO基底では収束が遅い
STO基底とGTO基底による結果は、それぞれの基底関数の規模を大きくしていく極限において一致します。しかし、STO基底がTZP規模ですでに収束しているのと比べると、GTO基底の収束が遅いのがわかります。彼らは、その要因として、原子核付近での正確な記述(カスプの再現)を可能にするSTOの優位性を指摘しています。
ECP基底では収束しない
ECP基底を使用した計算では、内殻電子の効果をポテンシャルに置き換えて計算しています。そのため、その結果がSTOやGTOなどの全電子基底の結果と一致するとは限りません。実際、ECP法に基づく計算では、かなり大きな基底関数系を用いた場合でもその結果が収束していないことがわかります。
このように、真の原子軌道の特徴(カスプと指数関数的な減衰)を再現するSTOを基底関数として採用することで、小さい規模の基底でも正しい結果が得られることが期待されます。
1) M. Güell et al., J. Phys. Chem. A, 112, 6384 (2008).
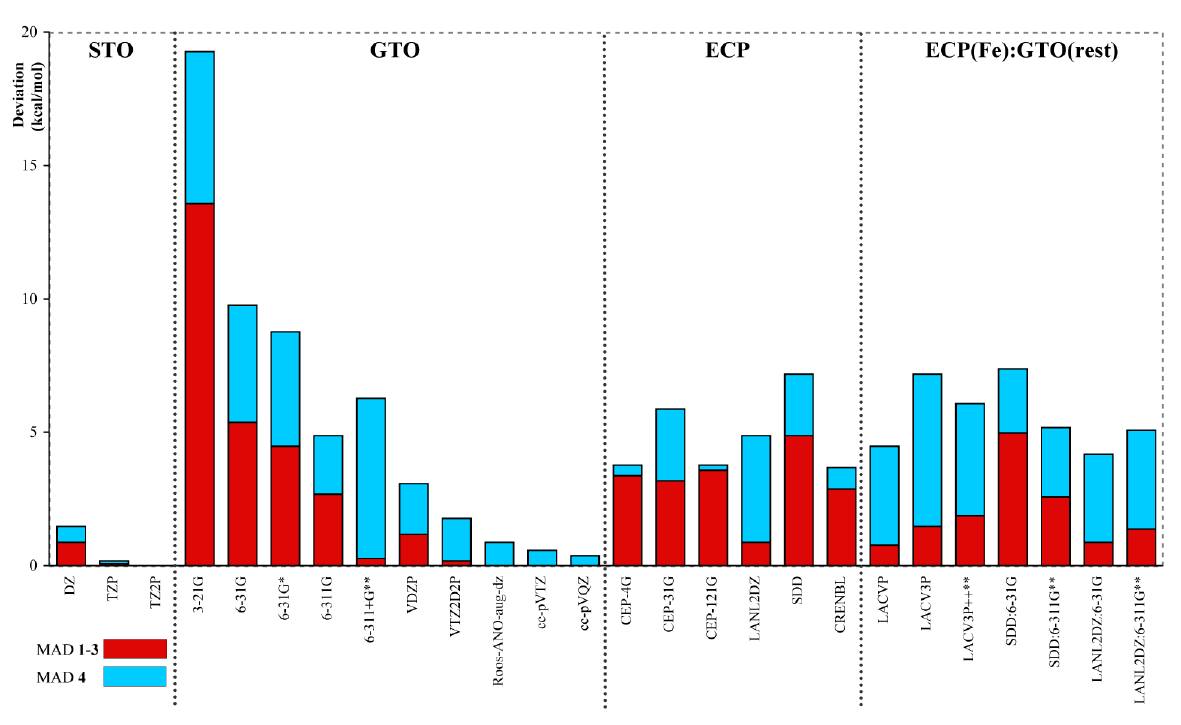
各基底関数の平均絶対誤差(kcal/mol)
title:{動作環境}
動作環境
以下の環境でご使用いただけます。
PC Linux (x86-64) 64-bit
MacOS (Apple Silicon) 64-bit
Windows (x86-64) 64-bit
並列スケーリング性能
SCM社は、主要なハードウェアメーカーと協力し、計算化学統合パッケージ「Amsterdam Modeling Suite(AMS)」が、あらゆるコンピュータ環境で最高の計算性能を発揮できるよう最適化を行っています。これには、様々なコンパイラやハードウェア構成に合わせたプログラムの微調整も含まれます。
彼らのチームは、最新のスーパーコンピュータ(HPC)環境において、計算規模を大きくしても性能が低下しないようにする「スケーリング性能」の向上に日々取り組んでいます。
以下は、AMSが高い並列計算性能を発揮する具体例です。
(使用マシン:32コア Intel Xeon Gold 6130 CPU @ 2.10GHz、メモリ192GB)
ゼオライト(496原子):ADFを使用したM06-L/TZPレベルでの力の一点計算
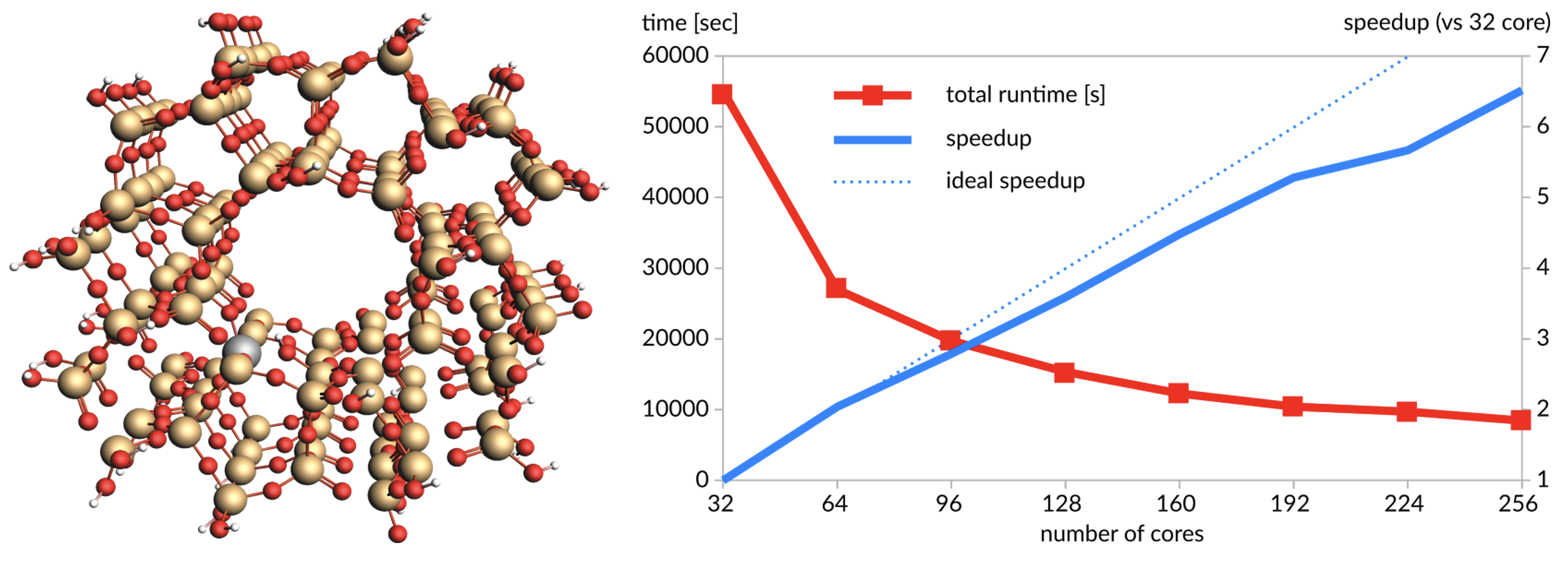
白金錯体(105原子):ADFを使用したBP86/TZ2Pレベルでの力の一点計算
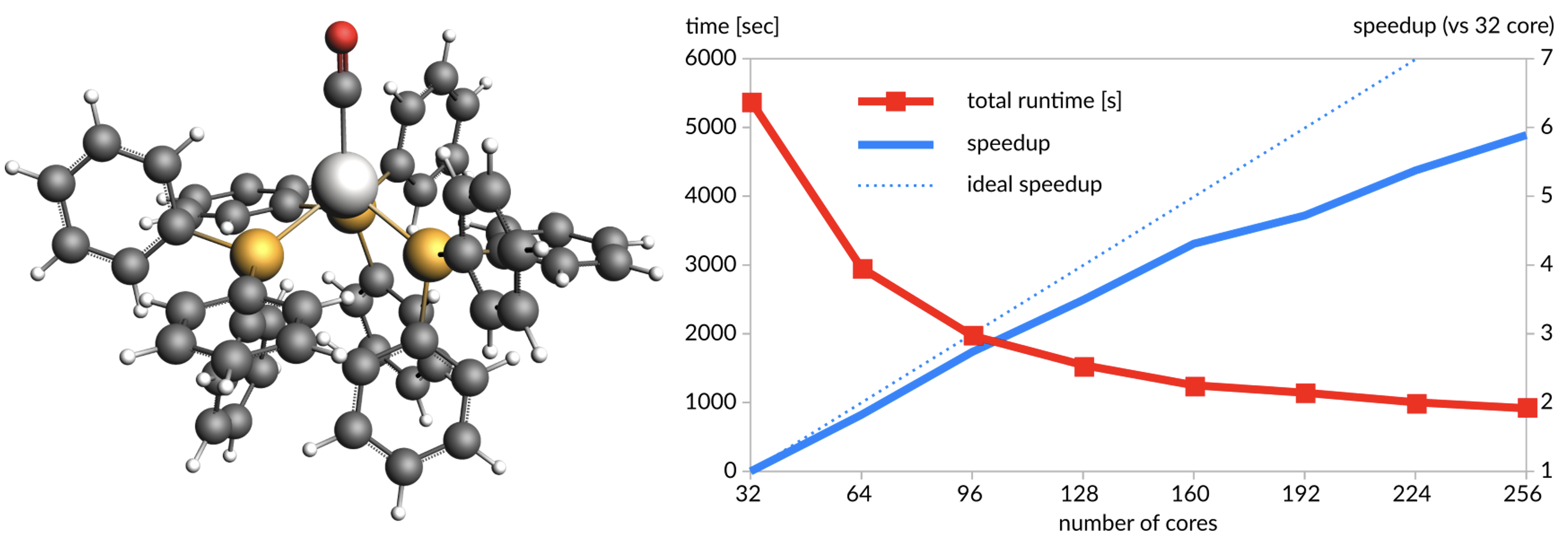
MOF結晶(468原子):BANDを使用したPBE/TZ2Pレベルでの一点計算
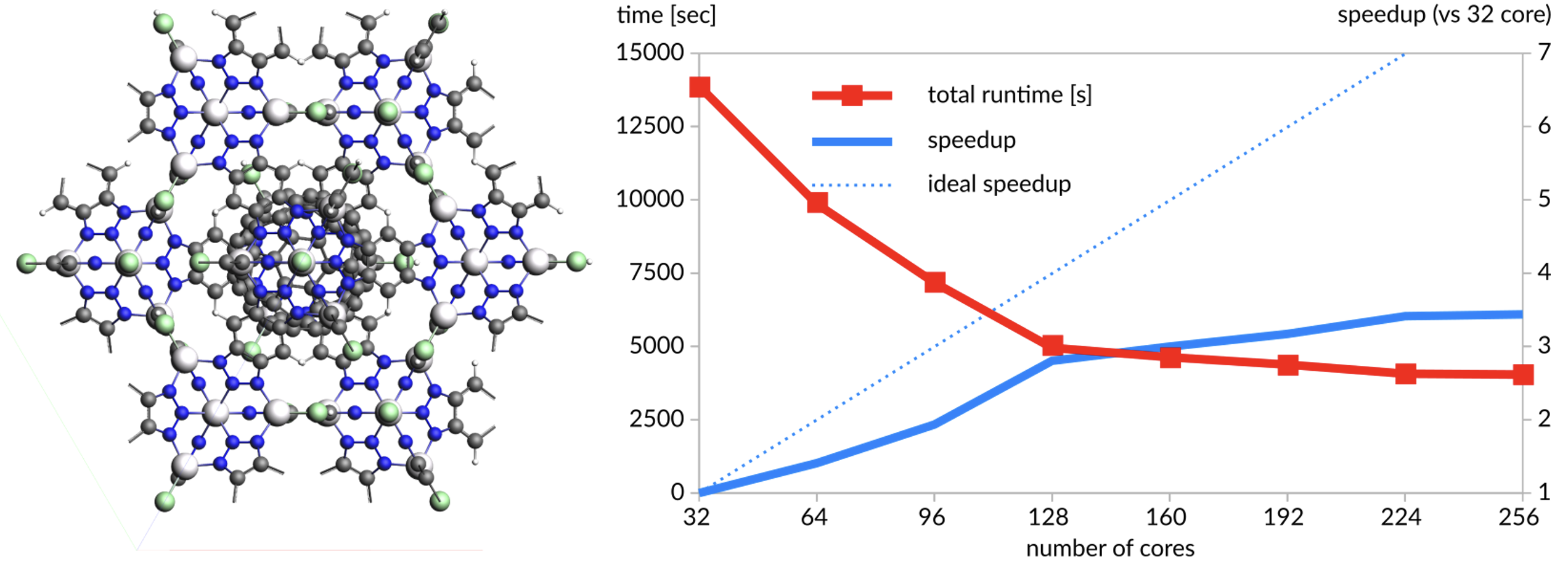
COF結晶(1440原子):SCC-DFTBを使用した一点計算(単一ノード内での比較)
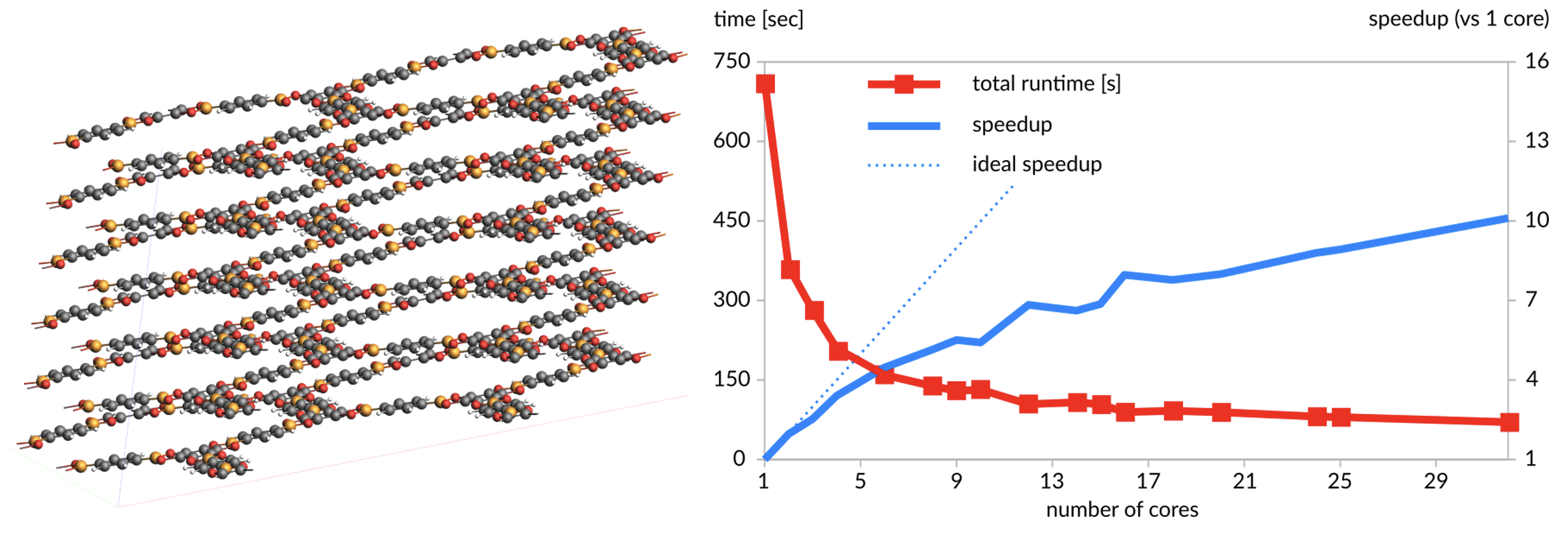
PETN(32,480原子):ReaxFF(AMS版をReaxAMSと表記)を使用した100ステップMDのLAMMPSとの速度比較
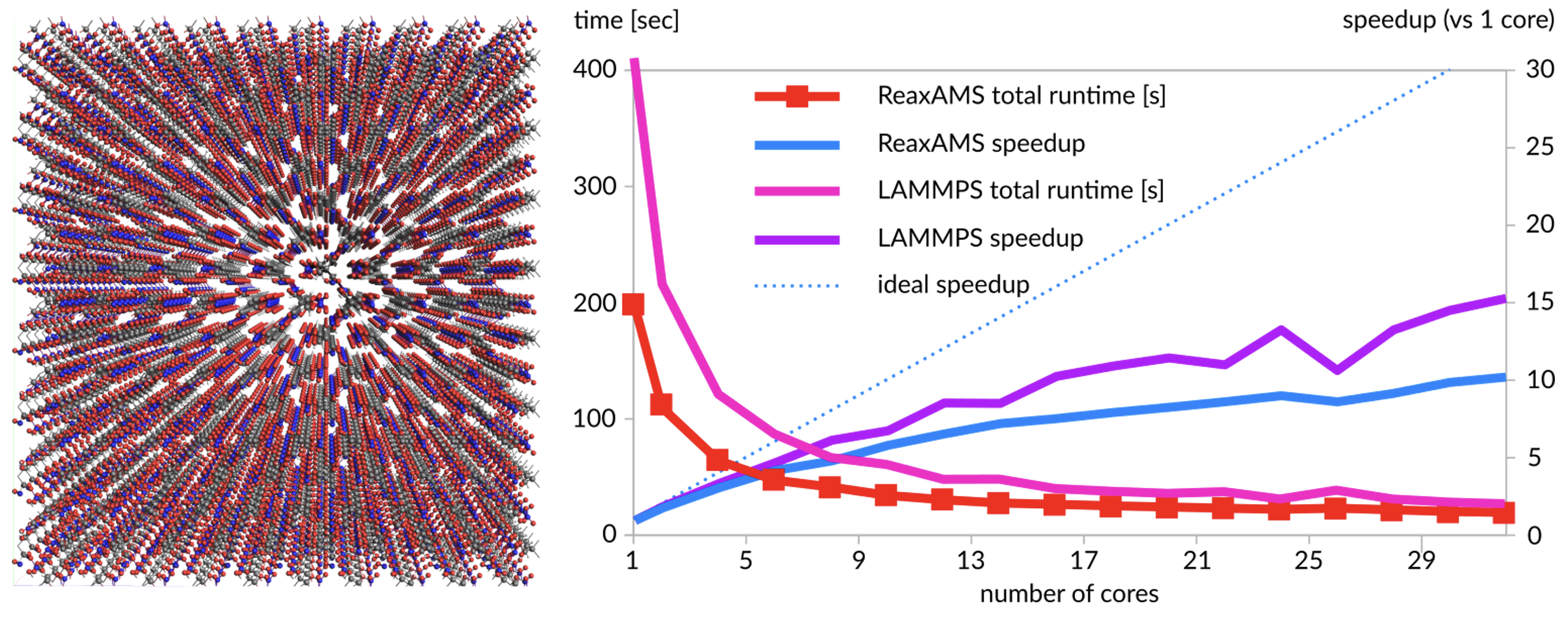
AMSの主要な計算エンジン(ADF, BAND, DFTB, ReaxFFなど)は、1台のマシン内の複数CPUコアを利用する場合(共有メモリ)と、複数台のマシンを連携させて計算する場合(分散メモリ)の両方で、効率的に並列計算できるよう設計されています。特にADFは、NMRや物性予測などの標準的な計算において、数百CPUコアを使用する規模まで性能が向上し続けます。
}:tab