


BioSolveIT社製ソフトウェア
(創薬支援ツール)
tab:{
title:{概要}
BioSolveIT社とは?
ドイツのBioSolveIT社は、創薬を視覚化するためのソフトウェアを開発しています。それらの高速で使いやすいソフトウェアを使用して、すべての化学者が研究を進めることができます。BioSolveIT社が開発したプラットフォーム「SeeSAR」と「infiniSee」は、受容体構造ベースおよびリガンド構造ベースの創薬を強力にサポートし、 分子設計における新しい鮮明なアイデアを創出します。

パンフレット

SeeSARパンフレットPDF版(0.8MB)
infiniSeeパンフレットPDF版(0.8MB)
FlexSパンフレットPDF版(0.5MB)
FlexXパンフレットPDF版(0.5MB)
FTreesパンフレットPDF版(0.8MB)
CoLibriパンフレットPDF版(0.3MB)
PoseViewパンフレットPDF版(0.4MB)
BioSolveIT社ウェブサイト
BioSolveIT社ウェブサイトもあわせてご覧ください。

YouTubeチャネル
BioSolveITTutorialsチャネルでは、BioSolveIT社創薬支援ツールの紹介、チュートリアル、ウェビナー、ワークショップ等を視聴いただけます。

title:{SeeSAR}
SeeSAR
SeeSARは、直感的で視覚的な医薬品設計プラットフォームです。
SeeSARは、バーチャルスクリーニングからフラグメント構造に基づく分子設計などの医薬品探索プロセスを幅広くをカバーし、化学者は楽しみながら包括的に新規リガンド設計を実施することが出来ます。
SeeSARの特長

Fast
迅速かつ効率的な計算により、化合物のドッキング、設計、分析を瞬時に行えます。

Visual
原子の色付けと美しい視覚化によって、リガンドと標的分子との相互作用を評価します。

Easy
オンザフライでの医薬品設計が可能です。
学習は必要ありません。
ユニバーサルな創薬ツールキット
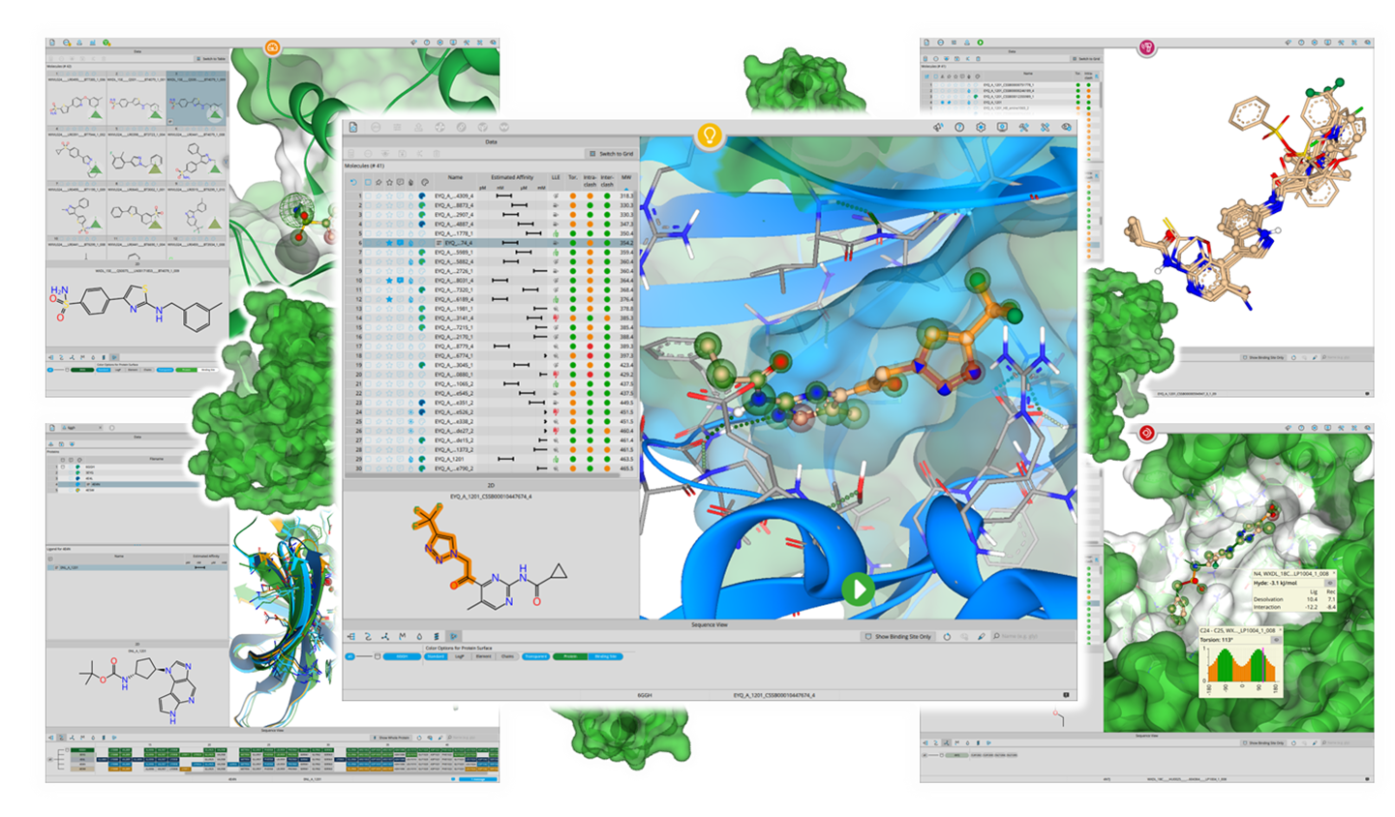
SeeSAR は、医薬品設計プロセスのあらゆる段階でのイノベーションを促進します。SeeSARには、化学者のニーズに合わせて微調整された、化合物と標的構造の処理に不可欠な様々なツールが含まれています。結合自由エネルギー寄与での色分け、結合ポケット中の非占有空間の可視化、ADME 特性計算などの有益な機能が、分子設計における意思決定をサポートします。BioSolveIT社のツールは、1,000 を超える出版物で引用されており、確固とした透明性のあるサイエンスに基づいています。
SeeSARに搭載されている各種モード

Protein Mode
構造ファイルをドラッグ&ドロップするか、データベースから検索することでタンパク質構造を読み込めます。数秒以内に分子構造が準備されます。

Protein Editor Mode
ニーズに応じてタンパク質構造を編集できます。回転異性体の探索や、変異の導入が可能です。

Binding Site Mode
SeeSAR は、リガンド結合部位を自動的に検出します。手動で残基を追加し結合部位を拡張したり、タンパク質内のポケット候補を探索することもできます。

Molecule Editor Mode
2D または 3D 構造を確認しながら、化合物を適宜修正できます。修正が完了すると、分子はリストに保存されます。

Analyzer Mode
視覚化された HYDE スコアを使用して親和性を推定し、結果を解釈します。関連するパラメータで化合物をフィルタリングし、ADME プロパティを計算し、リガンドとターゲットの相互作用を制御します。

Inspirator Mode
このモードでは化合物設計のアイデアを膨らませることができます。新しい母核を発見し、探索し、空洞に対して自由に伸張させたり、フラグメントライブラリを使用して分子を結合させたりして、より妥当な新規候補構造を構築します。

Docking Mode
1 回のクリックで化合物をドッキングします。ライブラリから活性化合物をスクリーニングし、直感的に結果を評価できます。
Space Docking Mode
Chemical Space Docking™ワークフローにより、超巨大なケミカルスペースの構造ベースのバーチャルスクリーニングを実施します。

Similarity Scanner
分子の類似性に基づいて、ターゲット構造を必要とせず化合物を整列させます。
フラグメントファイル
FastGrowで結合部位を満足させる
SeeSAR は、ターゲット構造の結合部位における空洞を探索し、それを埋めるためのリガンドの修飾またはフラグメント伸張を行い、高速にスクリーニングするための強力なツールを提供しています。この超高速コンポーネントである FastGrow は、形状ベースの方向記述子を使用した新規かつ効率的なアルゴリズムを採用し、標準的なハードウェア上で数秒以内に数十万のフラグメントをスクリーニングし、妥当な候補構造を提案します。
複数のフラグメントライブラリーが提供されています。
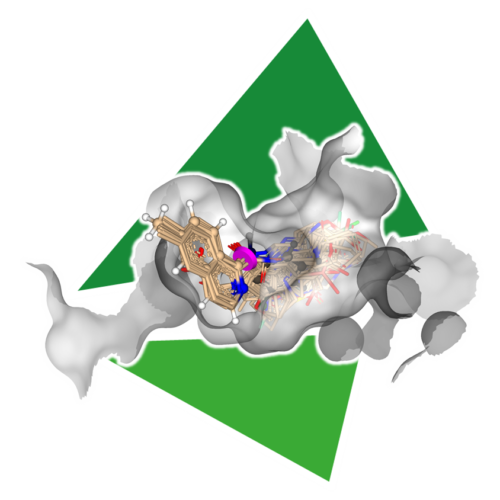
分子の3D構造を用いた母核置換
SeeSARは、新しい知的財産の創出や分子の問題の解決を支援します。SeeSARは、Inspiratorモードに実装されたReCoreアルゴリズムによって、選択した部分構造に適合する置換候補を、前処理済みのライブラリー、いわゆる「インデックス」から検索します。フラグメントベースのリード探索(FBLD)に対するBioSolveITのアプローチでは、母核置換、フラグメント伸張、フラグメント連結を数秒以内に処理します。
複数のインデックスファイルが提供されています。
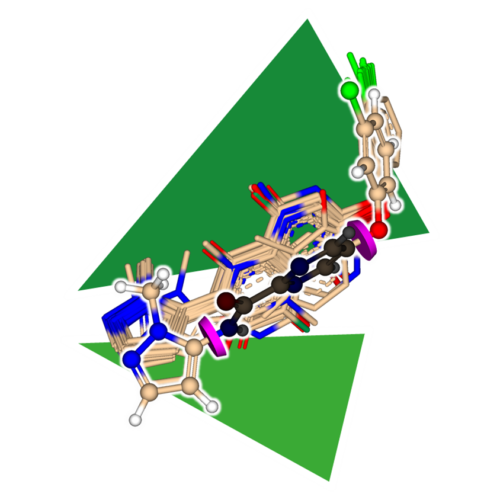
SeeSARに搭載されているアプリケーション
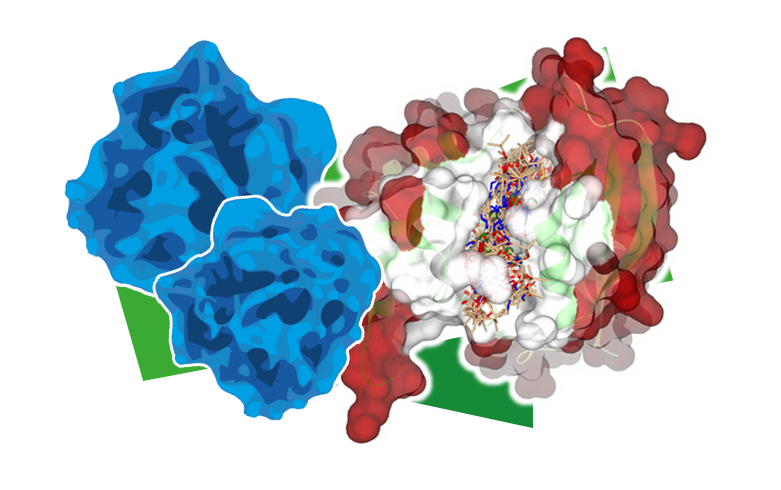
Chemical Space Docking™
Chemical Space Docking(C-S-D)™は、膨大なケミカルスペースをスクリーニングして最も有望な薬剤候補を特定する、新しい構造ベースのバーチャルスクリーニング手法です。
ユーザーはSeeSARの視覚的なインターフェースで、C-S-Dワークフローを簡単に開始することができます。計算開始後、HPSeeがその計算を引き継ぎ、結果を出力する準備を行います。
共結晶化リガンドやドッキングスタディから予測されたリガンドの結合ポーズをテンプレート分子として使用することで、シントンの初期配置を制限することができます。このアプローチは計算を高速化すると同時に、テンプレート分子と構造的に整合するドッキングポーズを生成します。
C-S-Dについてはこちらをご参照ください。
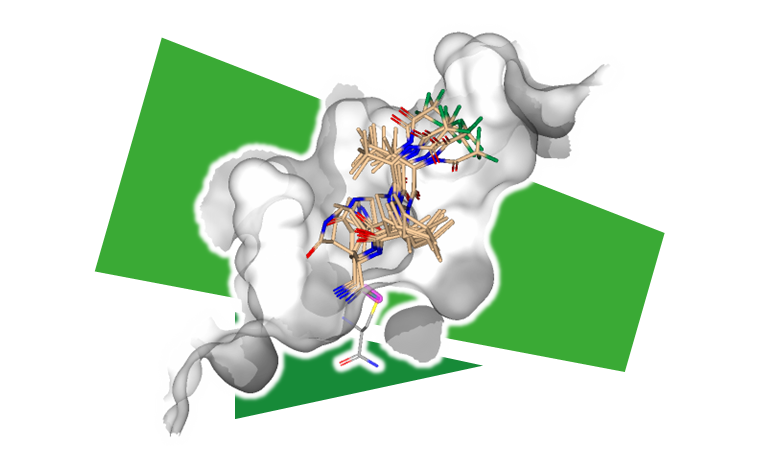
共有結合ドッキング
SeeSARの共有結合ドッキングは、提供されたリガンド内の共有結合弾頭を自動的に検出し、最も一般的に使用される 36 個の化学反応ルールを用いて、リガンドとターゲットタンパク質を結合させます。 SeeSARは、ポーズ生成時に形成された共有結合の柔軟性を考慮し、正確な出口ベクトルが既知の場合は剛体として保持することもできます。
さらに、共有結合のターゲットとして有効な任意の残基側鎖をユーザーインターフェースで選択し、サンプリングすることも可能です。
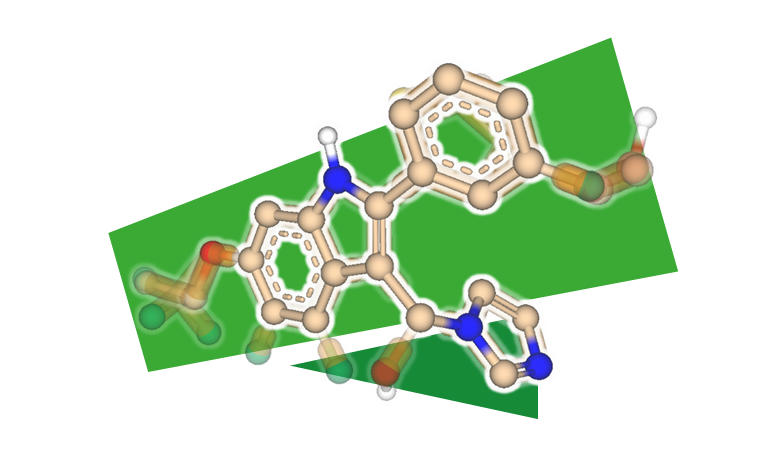
MedChemesis - リガンド変換ツール
MedChemesisは、薬化学で使用される一般的な変換ルールに基づいてリガンドを変換することができます。リガンドとタンパク質の複合体構造を入力として、様々な変換(例えば、メチル基の導入、カルボン酸のテトラゾールへの置換、芳香環の炭素原子と窒素原子の置換など)を行い有望な構造を提案します。
この方法の利点は、すべての位置におけるすべての可能な変換を列挙するのではなく、興味深い変換のみが生成されることです。例えば、表面との分子衝突につながる可能性のある置換基の導入は行いません。標準的なハードウェアを使用した場合、50件の提案を数秒行います。したがって、MedChemesisは、リガンド編集のためのアイデアを生み出し、その後の合成のための化合物の小セットを作成に利用できます。
MedChemesisは一般的に使用される290の化学反応を用いています。
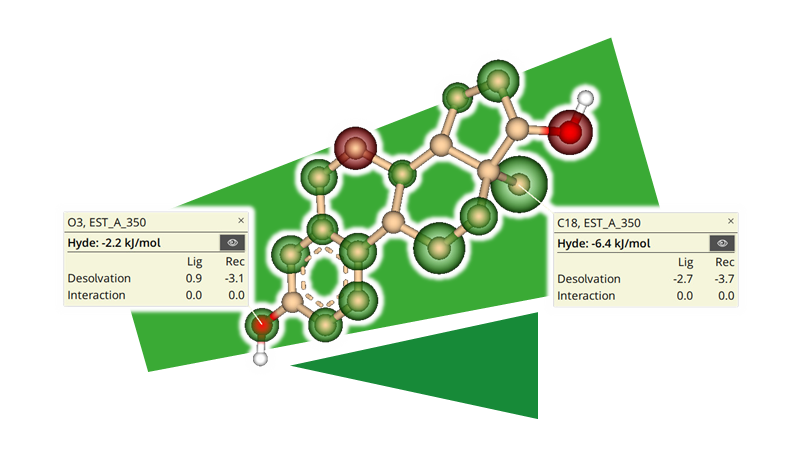
HYDE - インタラクティブで脱溶媒を考慮した視覚的な ΔG 推定
HYDEの結合評価は、親和性を近似し視覚化します。このシステムは特定のターゲットに対してトレーニングされたものではなく、代わりに、すべての力場に見られるような重み付けパラメータなしで、水素結合と脱水和をバランス良く評価します。ユーザーは、HYDEでの可視化により、リード最適化やその他の分子設計タスクについて、即座に解釈可能なフィードバックを得ることができます。 HYDEは、BAYER、ハンブルク大学、BioSolveITとのコラボレーションにより開発され、継続的に改良が加えられています。
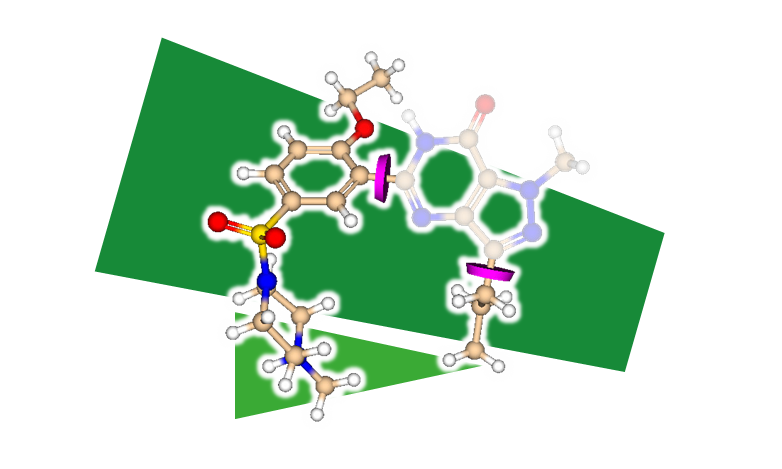
ReCore - 3D 母核置換
指定されたコアを置き換え、新しい知的財産を生成します。新しいフラグメントと一致する結合または相互作用を指定し探索できます。速度を向上するために事前に処理(「インデックス化」)されたフラグメントライブラリを利用できます。4次元ベクトルを使用して結果が取得され、適合の品質が評価されます。
ReCoreは、Roche Basel とハンブルク大学とのコラボレーションにより開発され、その後、BioSolveITによって大幅に強化および拡張されています。
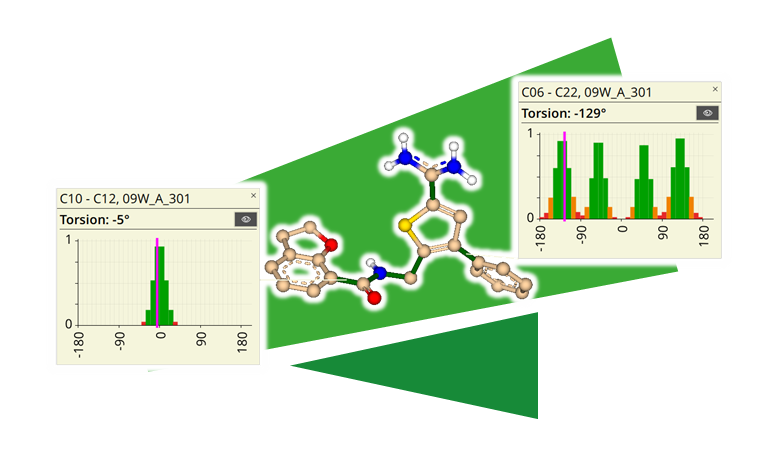
Visual Torsions - 二面角の可能性の統計的評価
結晶構造データベースの低分子の厳密な統計分析に基づいた、分子の二面角の「信号表示」は、特定の二面角の出現頻度を反映しています。基本的な仮定として、頻繁に出現する二面角は低エネルギー構造と相関し、その逆もまた同様です。Visual Torsionsは、Roche Baselとハンブルク大学とのコラボレーションにより開発されました。
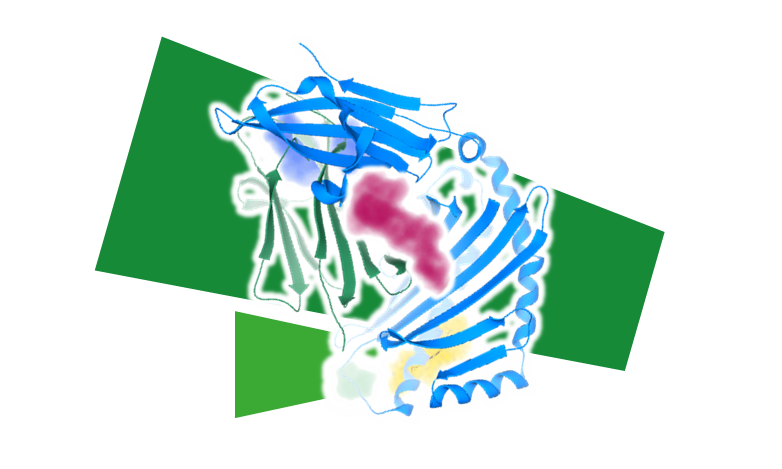
ポケット検出 - 3D構造から薬物結合部位を特定
アクセス可能な空のポケットの提案し、結果を 3D で視覚化し、解析を行うためのリガンド結合サイトとして選択します。この機能は、3Dグリッド上でガウス差分を使用するヒューリスティックモデルに基づいて、ポケットの形状を処理します。グローバルな水素結合と親油性特性、ポケットの溶媒接触表面(SAS)が考慮され計算されます。計算は、官能基原子のペア間の距離などのローカルな測定値によってさらに強化されます。
ポケット検出アルゴリズムは、Merck とハンブルク大学とのコラボレーションにより開発されたDoGSiteScorerが採用されています。

FlexXドッキング - 結合部位へのリガンドの高速かつ柔軟な配置
リガンドを受容体ポケットにドッキングします。この最先端のアルゴリズムは、リガンドをいわゆるフラグメントに分割し、ポケット内の複数の場所に配置し、シンプルでありながら非常に高速な事前スコアリング方式を使用してスコア付けします。配置されたn個のソリューションから、フラグメントごとにリガンドがさらに構築され、中間ソリューションは相互にスコア付けされます。そして、最も高いスコアを獲得したものがプロセスをパスし、ユーザーにドッキングポーズを提供します。「Single Interaction Scan」(SIS)配置は、化合物に極性基がほとんどない場合にもソリューションを見つけます。
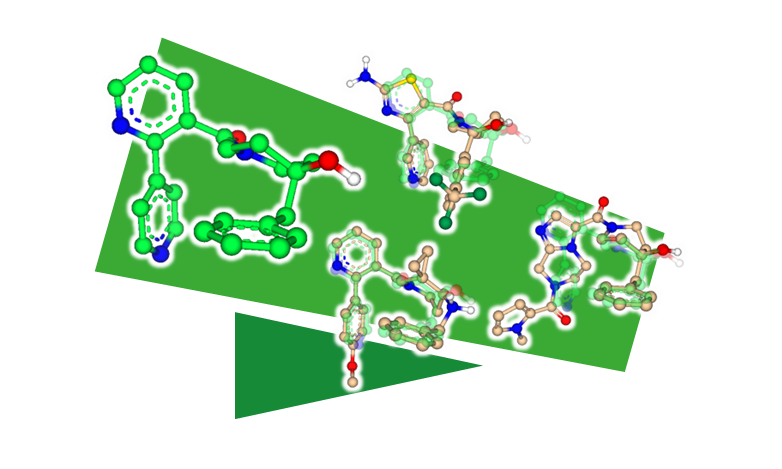
FlexS - リガンド構造に基づく類似性検索
FlexSはリガンドのアラインメント(重ね合わせ)を予測するツールです。FlexSは、与えられたリガンドのペアについて、一方のリガンドのもう一方に対する相対的な配座と配向を予測します。標的タンパク質の3D構造を必要とせずに、ユーザーはFlexSをSimilarity Scanner Modeの一部として使用し、分子セットの仮想スクリーニングを実行して、参照リガンドに類似した化合物を発見することができます。FlexSは分子の形状と分子特性を考慮し、母核置換や結合様式の仮説の生成に使用することができます。
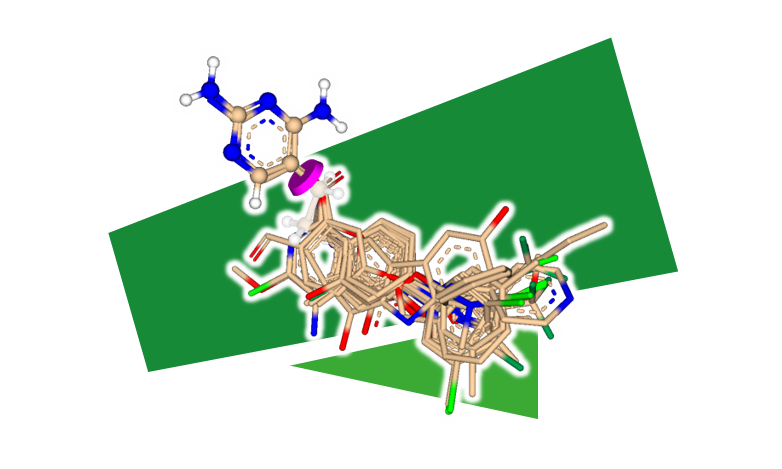
FastGrow — 超高速フラグメント伸張
FastGrow を使用すると、ユーザーは、標的タンパク質構造の空いている空洞を補完するリガンドの修飾またはフラグメント伸張を驚くほど高速に検索できます。このツールにより、インタラクティブな探索的伸張が可能になり、ユーザーは伸張プロセスを制御できます。さらに、ユーザーはファーマコフォア拘束を適用して、目的に合わせて検索を微調整できます。 ハンブルク大学、Servier Paris および BioSolveIT とのコラボレーションによって開発された FastGrow は、すでに AbbVie で最初の成功を収めています。さまざまなシナリオでの実際のフラグメント伸張/置換の場面で徹底的に検証されています。
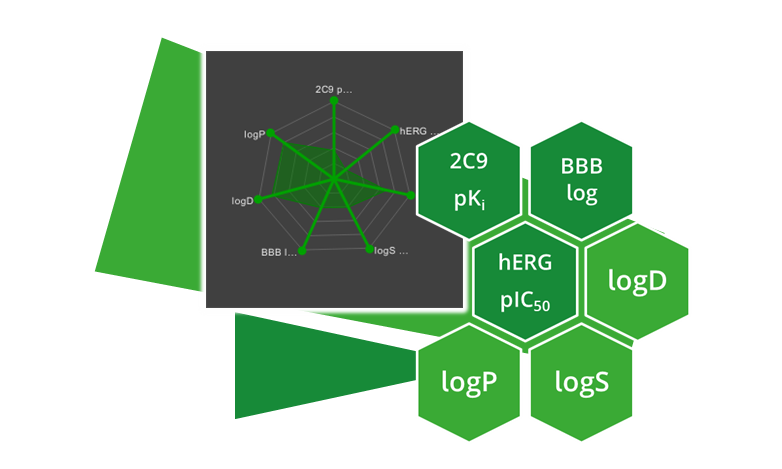
Optibrium - ADME 特性予測
物理化学的特性は、化合物が有望な薬剤候補であるかどうかを判断する際に考慮すべき重要な要素です。ADME パラメータを早期に評価することで、薬剤発見の初期段階で毒性の問題や吸収の課題を予測し、時間とリソースを節約できます。SeeSAR はさまざまな興味深いフィルタ パラメータ (回転結合、分子量、TPSA など) を計算できるだけでなく、Optibrium のオプションの StarDrop モジュールによる ADME パラメータ予測もサポートしています。これにより、ユーザーは CYP 酵素親和性、血液分布に対する血液脳関門、logS など、重要なパラメータを計算できるようになります。
YASARA によるエネルギー極小化計算
構造と複合体のエネルギー極小化は、バーチャルスクリーニング、ドッキング、SAR 解析の開始点を改善および改良するための基礎として役立ちます。ここでは、X 線結晶構造、モデル、または予測されたリガンド複合体を使用して、初期構造と比較してより安定な極小化構造(エネルギーレベルが低い配座)を計算します。YASARA モジュールは、SeeSAR のオプションです。
title:{infiniSee}
infiniSee
infiniSeeは、ケミカルスペース高速探索プラットフォームです。
プロジェクトのニーズに基づいて、ほぼ無限のサイズのケミカルスペースから類似性に基づいた関心のある分子を探索します。通常のアプローチでは、何百万ものエントリーを持つ列挙型の化合物ライブラリーの処理に数分から数時間を要することがありますが、infiniSeeでは、数秒以内に結果が得られ、購入可能な化合物や新規IPを提供します。
infiniSeeの特長

Fast
広大なケミカルスペースを
かつてないスピードで探索します。

Visual
直感的な色分けにより
類似性を一目で理解できます。

Easy
分かりやすいインターフェースです。
ドラッグするだけでクエリーの設定ができます。
infiniSeeの操作モード

Scaffold Hopper
クエリーとファーマコフォアが類似した構造的に異なる化合物を探索します。
FTreesアルゴリズムを使用しています。

Analog Hunter
フィンガープリントの類似性に基づいてクエリーと構造的に類似した化合物を探索します。
SpaceLightアルゴリズムを使用しています。

Motif Matcher
特定の部分構造を含む化合物、またはクエリー化合物と最大共通部分構造(MCS)を共有する化合物を探索します。
SpaceMACSアルゴリズムを使用しています。

Analyzer
ヒット化合物をまとめ、解析します。
Bemis-Murcko母核/骨格に基づいて分子をクラスタリングしたり、データをヒストグラムで可視化したり、フォローアップのための最も有望な候補をフィルタリングしたりできます。
3種類の探索モード
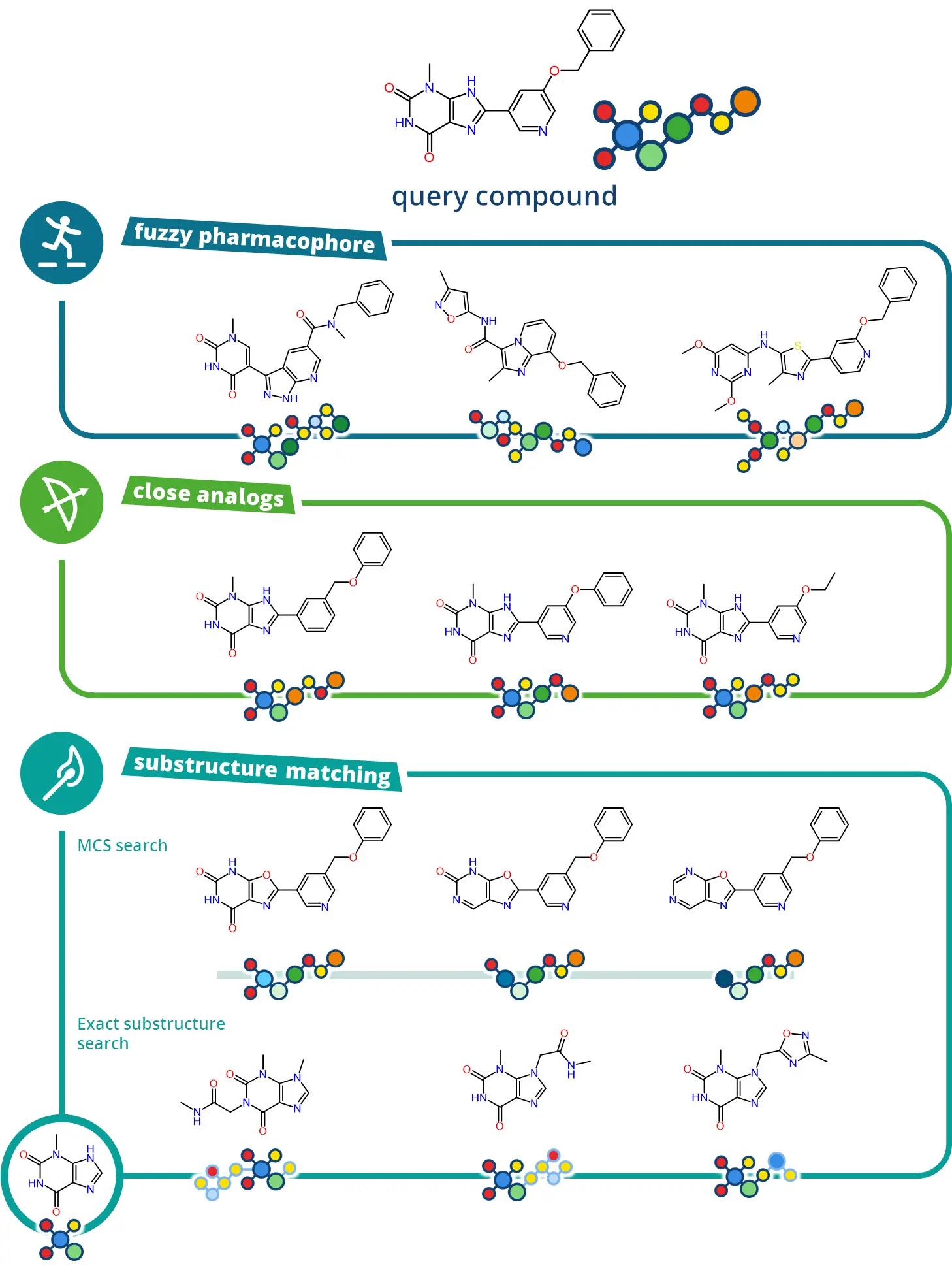
infiniSeeの様々な探索モードは、創薬の課題に応じてケミカルスペースから化合物を提供します。
Scaffold Hopperモードは、クエリーのファーマコフォアを維持しながら、新規性とIPポテンシャルの高い分子を提供します。
Analog Hunterモードは、クエリーの近傍のケミカルスペースを探索して、改善されたADME特性を持つシリーズ内のSARや具体的な分子バリアントを解明するのに役立ちます。
Motif Matcherモードは、2つの異なるアプローチを提供します: 1つ目のアプローチでは、クエリーと最大共通部分構造(MCS)を共有する化合物を提供します。2つ目のアプローチは、興味のある特定の部分構造を有する化合物を探索するもので、これによりフラグメント創薬(FBDD)の機会を解き放ちます。
最終的には、自分にとって何が重要かを決定し、様々な探索パラメーターや制約を適用しながらファインチューニングして最も関連性の高い化合物を得ることができます。
ヒット化合物の評価・解析

infiniSeeのAnalyzerモードでは、化合物データをまとめ、データに対する洞察を得ることができます。
Bemis-Murcko母核/骨格に基づいて化合物をクラスタリングし、各グループを調査して最も有望な候補化合物を選択できます。様々なフィルターを適用したり、ドラッグライク、フラグメントライク、リードライク化合物を直接検索したりすることもできます。
これにより化学的に多様な分子を高度かつ簡便に選択することができます。
化合物の各種パラメーターをヒストグラムにより可視化でき、データを掘り下げて分布の様々なセクション(例えば、MW、logP、水素結合ドナー/アクセプターの数)の調査に役立ちます。化合物に関連する試薬やビルディングブロックを調査したり、化合物をベンダーに基づいてグループ化したりする機能と組み合わせることで、ケミカルスペースを総合的に探索することができます。
利用可能なケミカルスペース
infiniSeeでは、以下のケミカルスペースから構造探索を行うことができます。
最新のケミカルスペースの情報は、開発元のウェブサイトをご覧ください。
table2:{ width:[20$20$20$20$20]$$
ケミカルスペース$化合物ベンダー$サイズ$購入可能<sup>※1)</sup>$合成可能$$
CHEMriya$OTAVA chemicals$1.2×10<sup>10</sup>$〇$ー$$
GalaXi$WuXi LabNetwork$1.2×10<sup>10</sup>$〇$ー$$
REAL Space$Enamine$7.0×10<sup>10</sup>$〇$ー$$
AMBrosia$AMBINTER$1.1×10<sup>11</sup>$〇$ー$$
Freedom Space$Chemspace$1.4×10<sup>11</sup>$〇$ー$$
eXplore$eMolecules$7.0×10<sup>12</sup>$〇$〇<sup>※2)</sup>$$
Knowledge Space$BioSolveIT$2.9×10<sup>14</sup>$ー$〇$$
}
※1: 化合物ベンダーにより合成され、数週間以内に入手可能。
※2: Cookbook (化学反応のリスト) を参照して、自社で合成可能。
ADME特性の予測
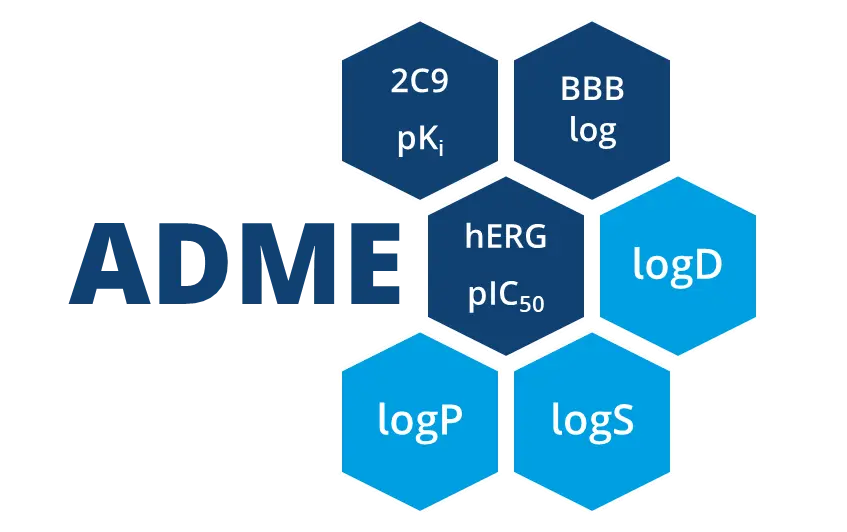
物理化学的特性は、化合物が有望な薬剤候補であるかどうかを判断するために考慮すべき重要な要素です。ADME特性も早期に評価することで、創薬の初期段階で毒性の問題や吸収の課題を予測し、時間とリソースを節約することができます。
infiniSeeは、様々な物理化学的特性(回転可能な結合数、分子量、TPSA等)を計算することができますが、オプションのOptibrium社のStarDropモジュールによるADME特性の予測もサポートしています。これにより、CYP酵素親和性、血液脳関門から血液分布、logSなどの重要な特性を計算することができます。
詳細は、開発元の資料(PDF)をご覧ください。
title:{infiniSee xREAL}
infiniSee xREAL
infiniSee xREALは、Enamine社最大のケミカルスペースxREAL Spaceを探索するためのプラットフォームです。
機密データを第三者に開示することなく、自身のハードウェア上で数兆もの化合物コレクションに対して直接、高速かつ効率的なスクリーニングを行うことができます。
BioSolveIT社とEnamine社とのコラボレーション
infiniSee xREALは、BioSolveIT社とEnamine社の長年にわたるコラボレーションにより開発されたものです。早期の創薬フォローアップを加速させるために、超巨大ケミカルスペースxREAL Spaceをスクリーニングし、興味のある分子を探し出すことができます。
BioSolveIT社は、巨大ケミカルスペースを効率的にスクリーニングするための強力な技術を開発したパイオニアです。現代の創薬課題に対応するために、長年にわたり、関連する化合物を探索する様々な探索手法を開発してきました。
Enamine社は世界的に知られ、信頼されている化合物ベンダーであり、農薬や創薬研究のために化学的に多様な分子へのアクセスを提供しています。目的の化合物を数週間以内にお届けし、競争力のある価格設定を実現しています。
infiniSee xREALによる化合物探索

infiniSee xREALは、xREAL Space専用のinfiniSeeです。
従来のinfiniSeeと同様、数兆サイズのケミカルスペースから最も関連性の高い化合物を探索・解析するための操作モードを備えています:
- ニーズに応じて化合物を探索するための様々な探索モード:
- Scaffold Hopper: ファーマコフォアが類似した構造的に異なる化合物を探索
- Analog Hunter: 分子フィンガープリントに基づいた構造的に類似した化合物を探索
- Motif Matcher: 共通の部分構造、および最大共通部分構造(MCS)を持つ化合物を探索
- ヒット化合物を解析するためのAnalyzerモード:
- 検索結果の閲覧とフィルタリング
- 特性のヒストグラム表示による結果の解析
- Bemis-Murcko母核/骨格に基づくクラスタリング
- OptibriumモジュールによるADME特性の予測
xREAL Spaceについて
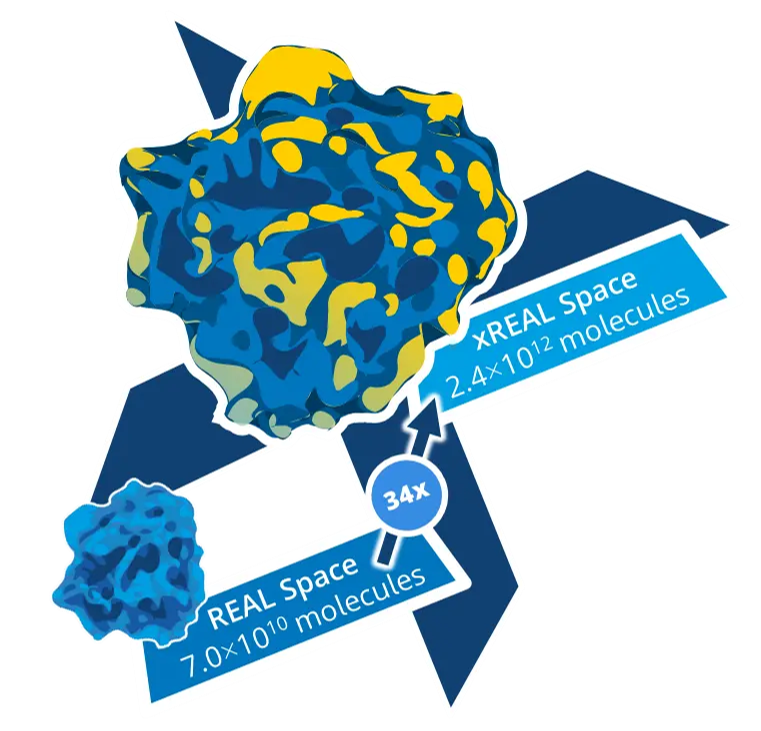
xREAL Spaceは、3,125種の化学反応と15.2万種のビルディングブロックの組み合わせに由来する2.4兆化合物からなる超大規模なケミカルスペースで、これは従来のREAL Spaceの34倍の大きさです。
Enamine社の広範なビルディングブロックと化学反応に由来する比類のない化学的多様性により、xREAL Spaceはさらに80%を超える高い合成成功率を保証しています。このケミカルスペースは、予測精度を高めるための機械学習アプローチによって常に改善されています。
xREAL Spaceからヒットした化合物は、従来のREAL Spaceと同様、注文するとEnamine社で合成され、数週間で入手することができます。
title:{HPSee}
HPSee
HPSeeは、ハイパフォーマンス・コンピューティングへのシームレスなアクセスを可能にします。
ジョブを調整し、可能性を拡大し、ワークフローを最適化して、妥協のない結果を快適な時間枠で達成します。
HPSeeの特長
Screen faster and smarter
コンピューティング能力を活用し、バーチャルスクリーニングデータに便利にアクセスできます。

Upscale your possibilities
広範な計算をリモートハードウェア上でスケーラブル実行できます。

Optimize your workflows
妥当な時間内で、妥協しない高い品質の結果を生み出せます。
ハイパフォーマンス・コンピューティングを強化

HPSeeは、効率的なバーチャルスクリーニング・ワークフローを実行するためのプラットフォームです。HPSeeは、標的構造へのドッキングなどの計算創薬のシナリオにおいて、大規模な化合物コレクションを容易に扱うことを可能にします。ユーザーは、SBDD統合プラットフォームSeeSAR上で計算のセットアップを容易に行った後、HPSeeを設定したハードウェア上で利用可能なリソースを活用してスムーズに計算を開始できます。計算が終了すると、ユーザーはその結果にアクセスし、さらに評価することができます。ライブラリーやデータの複雑な準備は必要ありません!クリーンな管理ダッシュボードは、直感的な管理インターフェースを備えています。管理者は、このプラットフォームを操作しながら、ユーザーのアクセス許可とともに化合物ライブラリーとケミカルスペースを簡単に監督することができます。
ドッキング計算を次のレベルへ

計算的手法、特に大規模なバーチャルスクリーニングは、低分子創薬の初期段階においてその重要性を確立しています。そのため、この手法への普遍的なアクセスを確保することで、結果とその後の個々のタスクの処理を迅速化し、プロジェクトのパフォーマンスを向上させることができます。
HPSeeを使用することで、バーチャルスクリーニングは誰にとっても身近なものになります。計算が初心者の方でも、SeeSARのグラフィカルなインターフェースから、複雑な準備をすることなく、簡単にスクリーニングキャンペーンを開始することができます。
リモートドッキング

HPSeeは、大規模な構造ベースのバーチャルスクリーニング・キャンペーンをサポートします。 化合物ライブラリーをHPSeeにアップロードし、SeeSARのリモートドッキングモードでライブラリーを選択します。計算が開始されると、HPSeeはHYDEを用いてリガンド-ターゲット複合体のポーズ生成とスコアリングを実行します。 最後のステップでは、HPSeeが結果を処理し、SeeSARで視覚的に確認できるようにします。
Chemical Space Docking™

HPSeeは、Chemical Space Docking™(C-S-D)を容易にするために開発されています。C-S-Dは、超大規模なケミカルスペースをスクリーニングし、最も有望な医薬品候補を探索する新しい構造ベースのバーチャルスクリーニング手法です。
ユーザーは、SeeSARの美麗なインターフェースで、C-S-Dのワークフローを簡単に開始することができます。その後、HPSeeが計算と結果の作成を引き継ぎます。
共結晶リガンドやドッキング計算により予測された結合ポーズをテンプレートとして使用することで、最初のsynthonのanchoringステップを強化することができます。このアプローチにより、テンプレート分子と構造的にアラインメントされたポーズを生成しながら、計算を加速することができます。
HPSeeによる恩恵

IT/管理者
チームの計算リソースへのアクセスを洗練された方法で管理できます。
HPSeeは、必要な要件を満たす柔軟性と拡張性を備えて開発されています。

メディシナルケミスト
豊富な専門知識がなくても、多数の化合物のドッキング計算を簡単に実行できます。

計算化学者
バーチャルスクリーニング・キャンペーンを効率化し、不必要なデータ操作から解放されます。
title:{Chemical Space Docking™}
Chemical Space Docking™
Chemical Space Docking™は、数十億を超える化合物の構造ベースの探索へのBioSolveIT社の対応策です。この斬新なアプローチは、卓越した効率と洗練された方法で最良の医薬品候補を掘り出します。
化合物の大宇宙からの関連する化合物の抽出
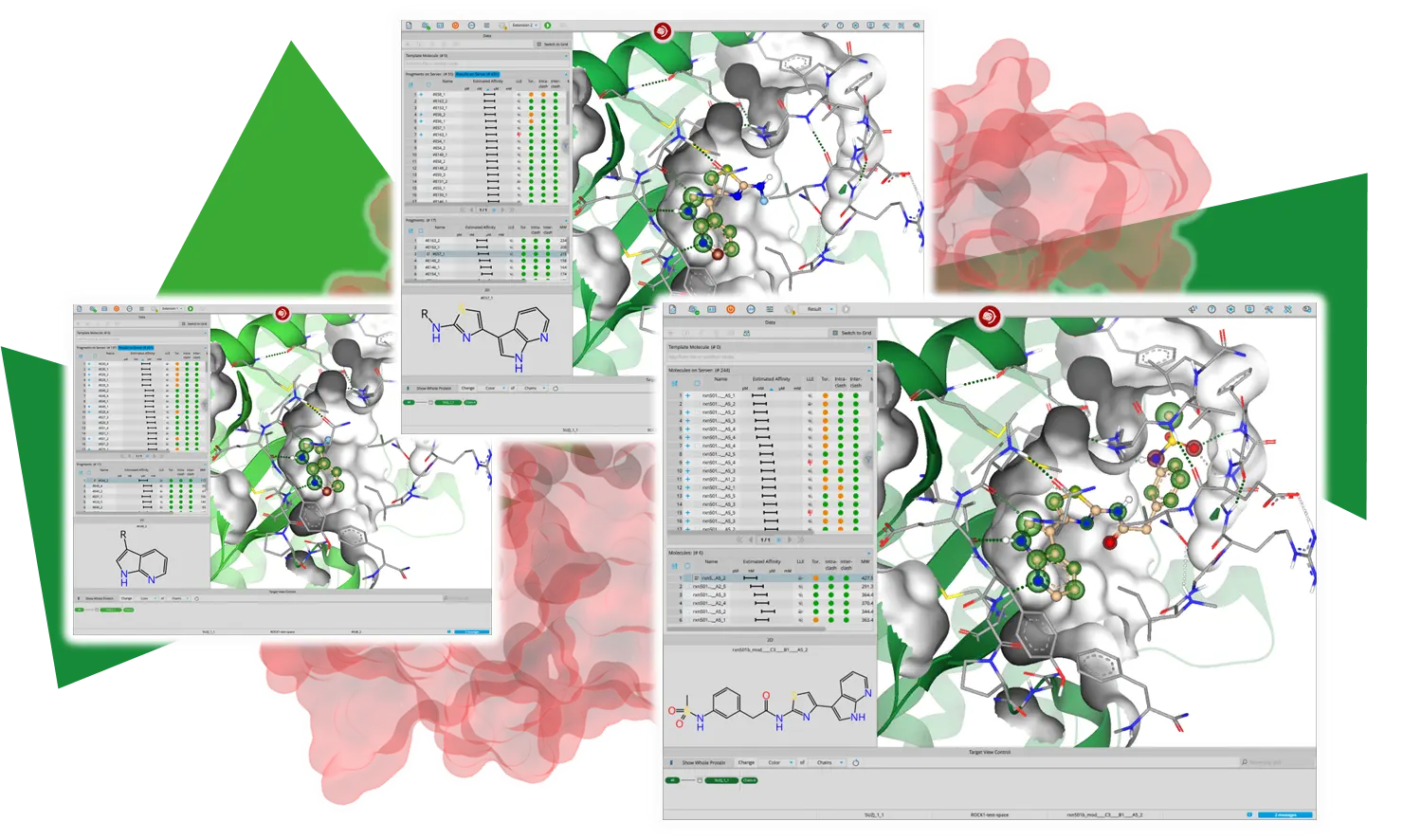
Chemical Space Docking™は、数十億あるいはそれ以上の化合物を包含する超大規模なケミカルスペースへのアクセスを提供します。
Chemical Space Docking™は、従来のバーチャルスクリーニングで一般的に採用されている、リソースを大量に消費する化合物の総当りのドッキングに頼る代わりに、結合部位に効果的に伸長する最も有望な候補化合物を優先的に選択します。
得られた結果は、合成可能性が高く、商用ケミカルスペースから得られたものは市販もされています。
ユーザーは、インタラクティブなワークフローにより、すべての段階で計算結果に関与することができ、プロジェクトを効果的に成功へと導くことができます。
Chemical Space Docking™のコンセプト
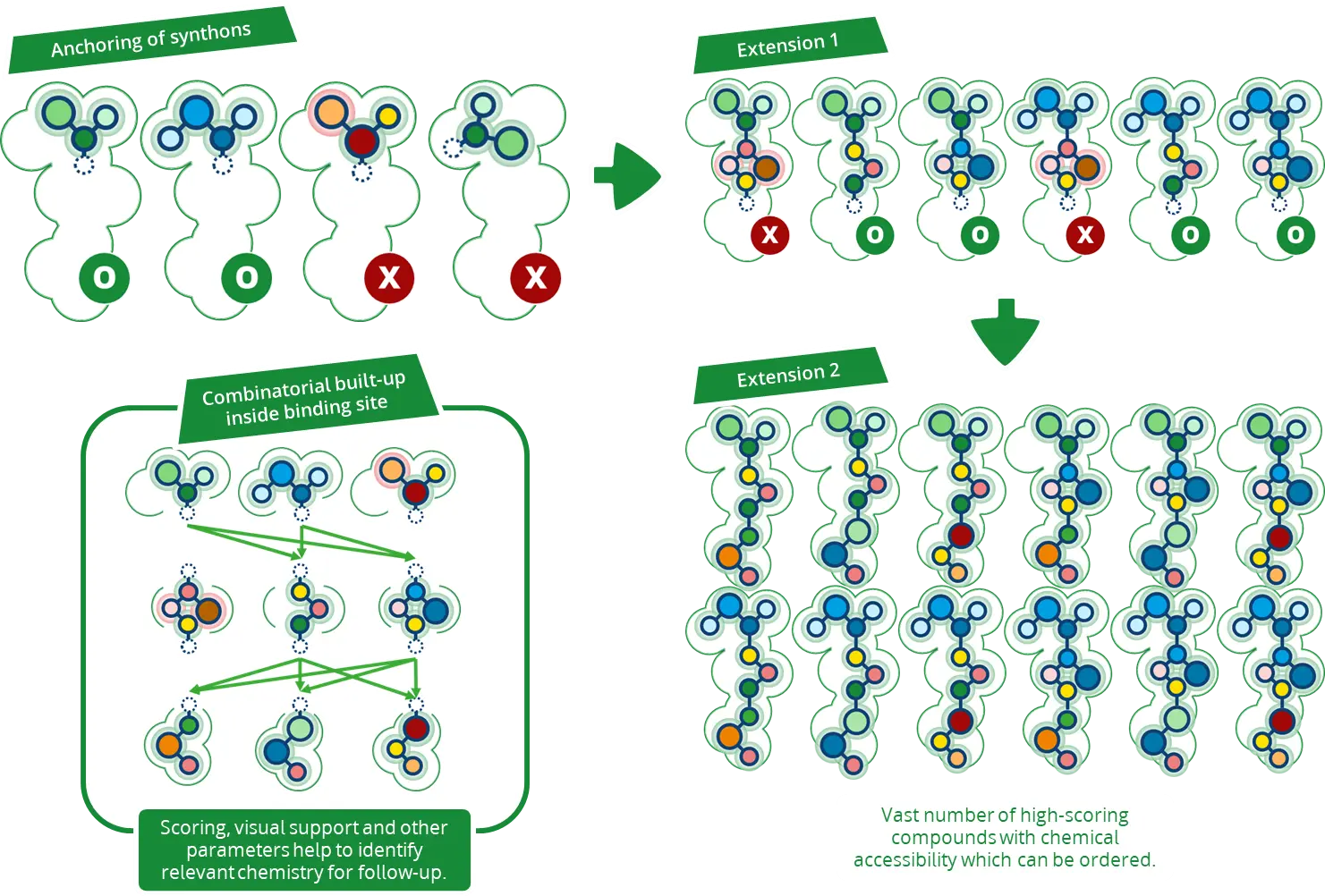
Chemical Space Docking™のコンセプトは、膨大な化合物コレクションを効率的に構造ベースで探索することです。
Chemical Space Docking™は、数十億を超える化合物を総当たりでドッキングするような数ヶ月から数年かかる可能性のある手法ではなく、有望で関連性の高い化合物にのみ焦点を当て、計算時間とリソースを節約します。パフォーマンスの低い薬剤候補は、ワークフローの初期の段階で却下され、興味深い選択肢のみに焦点が当てられます。
医薬品候補を生み出す最小の特徴、すなわちビルディングブロックから出発して、最終生成物の化合物が結合部位内でコンビナトリアルに組み立てられます。定義された化学反応ルールを用いることで、得られた化合物は合成可能性が高く、また、BioSolveIT社が提携している化合物ベンダーを通じて購入することもできます。
最初のステップ: Synthonのドッキング
ワークフローは、選択されたケミカルスペースの作成に使用されているビルディングブロックのドッキングから始まります。
このステップでは、定義した結合部位にファーマコフォア拘束を適用することができます。これにより、synthon(化合物の最初の開始フラグメント)の配置がガイドされ、ターゲットと望ましい相互作用を形成できるものが特定されます。
Synthonのドッキングが完了すると、SeeSARにより、生成されたポーズを視覚的に調べることができます。それぞれのsynthonには、「リンカー原子(linker atom)」と呼ばれる伸長方向を示すマーカーが付けられているため、評価プロセスを簡素化できます。
選ばれた候補化合物は、ワークフローの次の段階である伸長ステップに進みます。
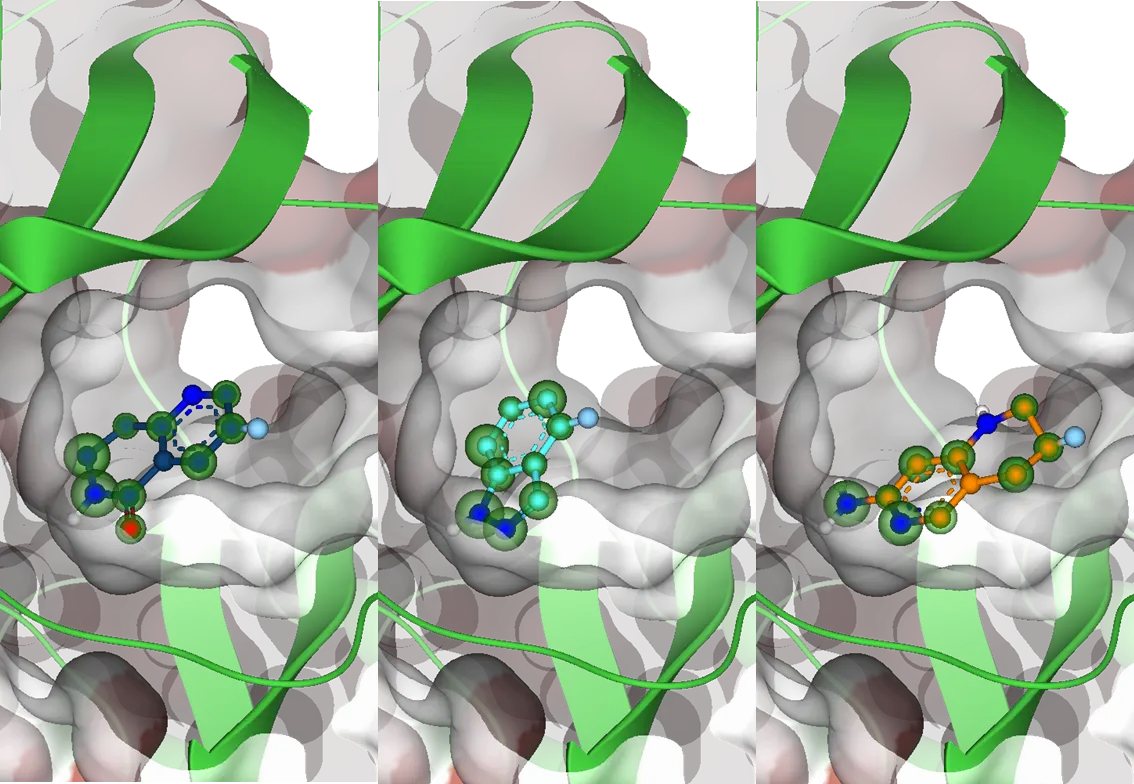
Synthonの伸長: 医薬品候補への進化
Synthonのドッキングステップの後、選択されたsynthonは、伸長ステップでドラッグライクな化合物へと拡張します。ここでは、最初のフラグメントは、ケミカルスペースの化学反応ルールに従って、他のビルディングブロックと組み合わされます。合成可能な化合物のみが作成され、結合部位での相互作用が評価されます。
SeeSARでのセットアップにより、伸長した化合物のドッキングを素早く正確に処理し、synthonの出発点の結合様式を確実に反映させます。このアプローチは、従来のドッキング手法よりも数倍速く、進化した化合物が出発フラグメントの方向と一致することを保証します。
ここでも、ユーザーは、結果に追加の相互作用パターンを導入するためにファーマコフォア制約を使用してプロセスを導くことができます。
計算が終わると、2種類の結果が表示されます: リンカー原子のない化合物と、リンカー原子のあるの化合物です。リンカー原子を含まない化合物は最終分子であり、化合物ベンダーに注文、あるいは合成することができます。リンカー原子をまだ含む化合物は、最終分子につながる追加の伸長ステップの計算が必要です。
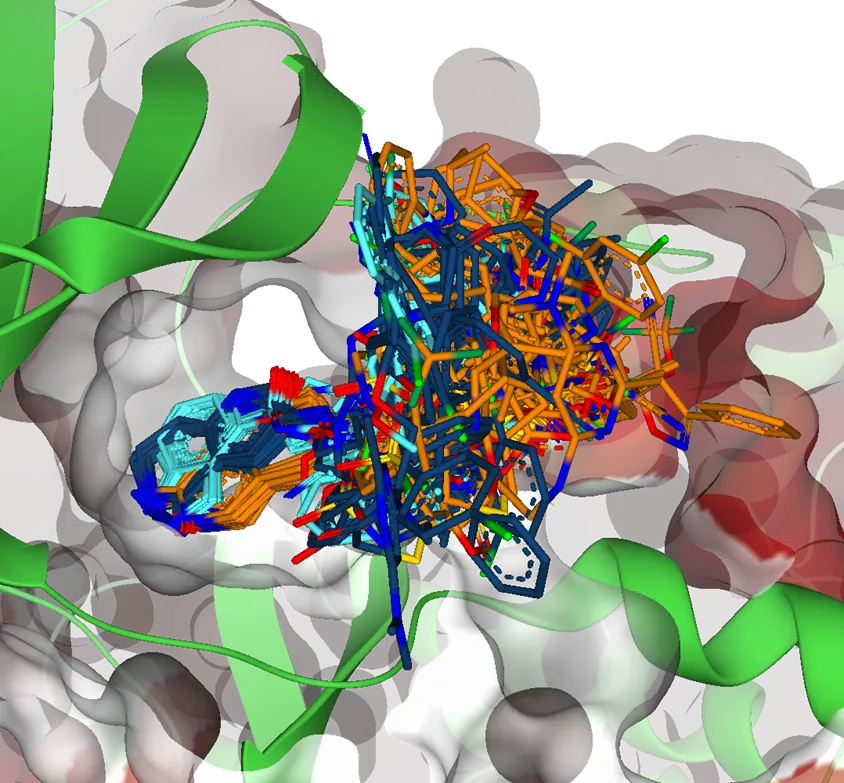
ランダムドッキングよりもスマートな手法
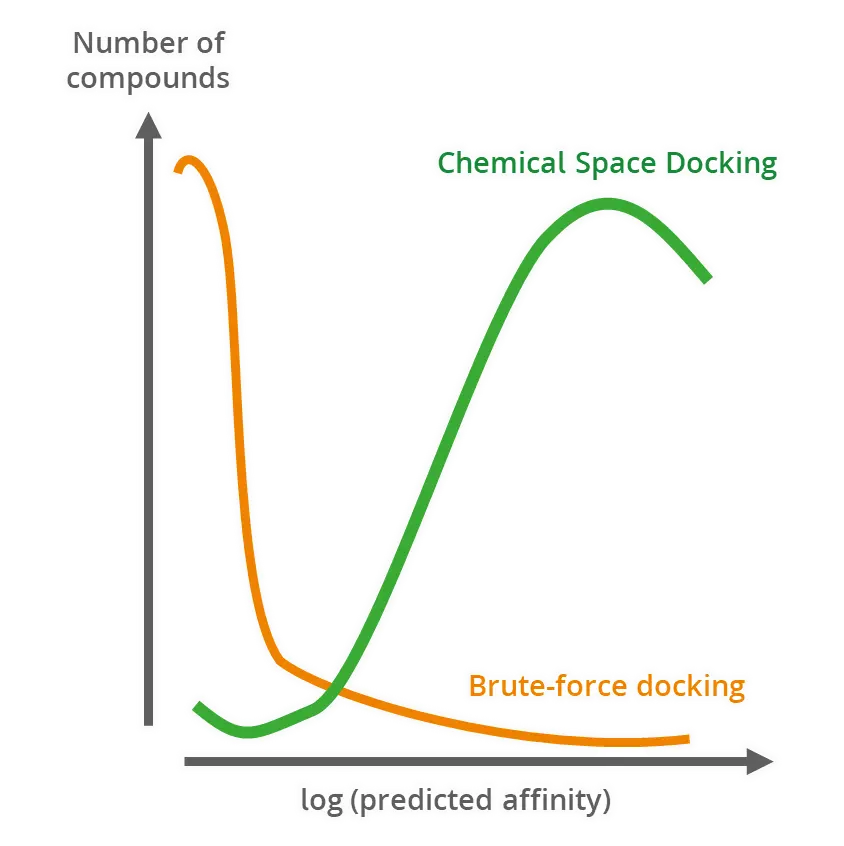
分子のランダムなサンプリングでは、ほとんどのケースにおいて95%以上のリガンドについてひどいドッキングスコアになることが実証されています。
このことは、本質的に、従来のバーチャルなハイスループットスクリーニングでは、計算のかなりの部分が根本的に不適当な候補に割り当てられていることを意味します。
総当たりのドッキング(Brute-force docking)手法と比較すると、Chemical Space Docking™はプロセスを大幅に加速し、効率的に高スコアの化合物の多様なセットをわずかな時間で濃縮します。
大胆に言えば、より多くの優れた化合物をより速いペースで発見することができます。
Chemical Space Docking™の利点
多くの時間の節約
何十億もの化合物のドッキングは、標準的なハードウェアでは数ヶ月から数年かかることがあります。Chemical Space Docking™は、同じことを数日で行います―ほんのわずかな時間です!

インタラクティブなワークフロー
ターゲットはあなたが一番よく知っています。ファーマコフォア拘束を適用することで、その知識をスクリーニングの指針として活用できます。さらに、SeeSARは、ターゲットとリガンドの複合体を理解するための視覚的なサポートを提供することで、フォローアップのための最良の候補を選び出すのに役立ちます。
様々なケミカルスペースが利用可能
BioSolveIT社と化合物ベンダーとのコラボレーションにより、さまざまな商用ケミカルスペースが利用できます。いずれもユニークな化合物を生み出すためのユニークなビルディングブロックとインハウスの化学反応を備えています。
洗練されたユーザーインターフェース
SeeSARは、Fast、Visual、Easyをコンセプトに設計されています。Chemical Space Docking™ワークフローの各段階は、理解しやすく、操作も容易です。これにより、計算経験の浅いメディシナルケミストや創薬の初心者でも、新規IPや有望な結果を得るために超大規模な化合物コレクションを掘り下げることができます。

標準的なハードウェアで実行可能
大規模な計算リソースは必要ありません。SeeSARとChemical Space Docking™はセットアップが簡単であるため、小規模な企業や研究グループでも利用できます。

分子の新規性を取得可能
Chemical Space Docking™は、ケミカルスペースから新規で強力なケモタイプを探索することができます。ターゲットの様々なサブポケットを補完する多様な化合物を検索することができ、パイプラインに様々なスキャフォールドを提供できます。
title:{Chemical Space}
Chemical Space
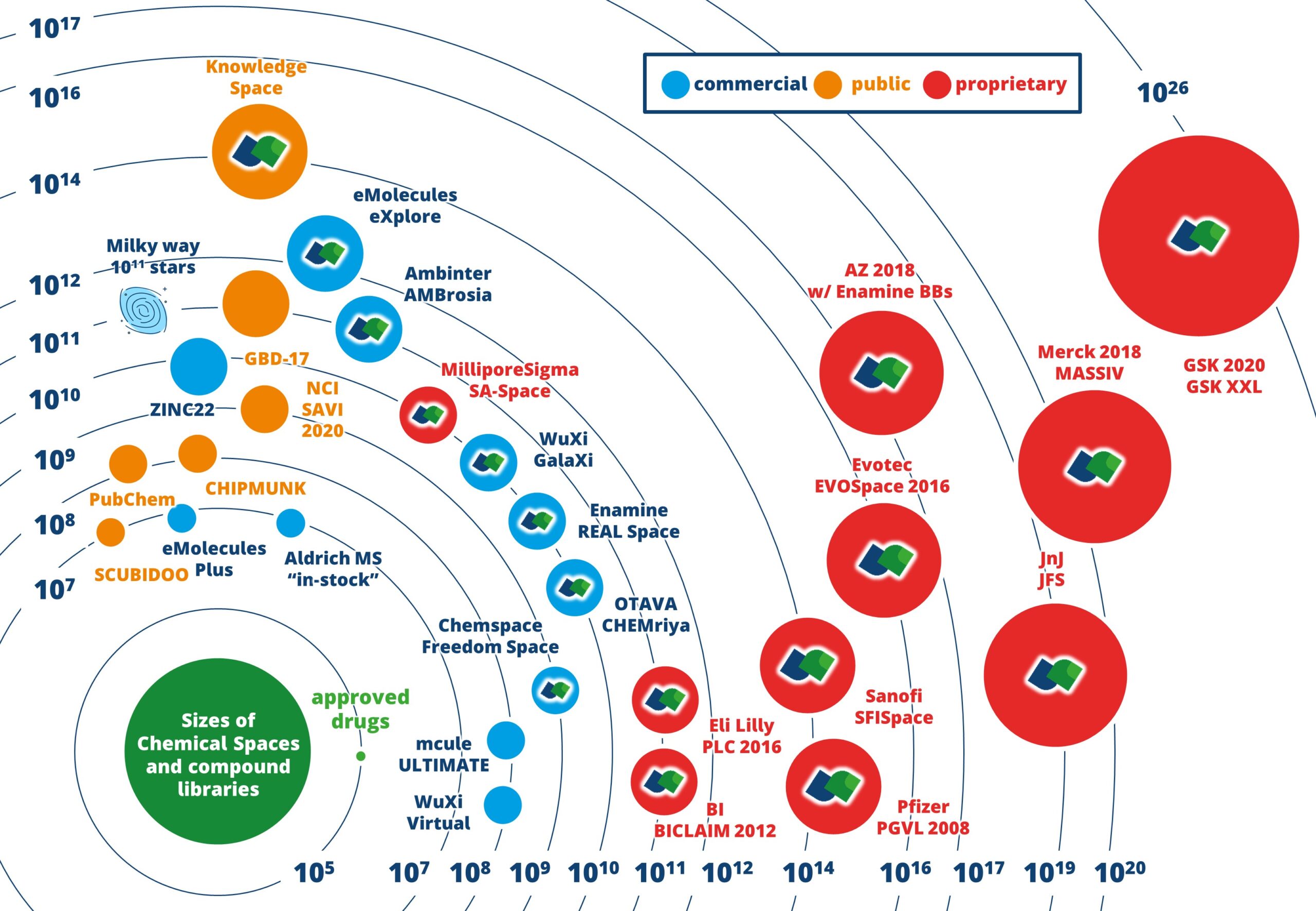
次世代の大規模分子コレクション
BioSolveIT は、個々のケミカルスペースの開発と探索の先駆者です。ケミカルスペースとは、合理的な時間枠内で効率的にスクリーニングできる数十億~数兆のエントリを含む分子コレクションを指します。ケミカルスペースは、ビルディングブロックと定義済みの化学反応ルールを組み合わせることで作成されます。列挙された分子ライブラリとは異なり、エントリは明示的にリストされていません。その代わりに、組み合わせ論によるケミカルスペースでは、化合物を高速かつ動的に生成して迅速な処理を可能にし、合成可能な化合物あるいは購入可能な化合物を提供します (ケミカルスペースによって異なります)。
組み合わせの驚異

ケミカルスペースは、分子のセットを網羅的に列挙するという制約を超え、すべてのエントリをカバーします。
ケミカルスペースは、ビルディングブロックとそれらを組み合わせる方法に関する化学反応ルールという 2 つの要素で構成されています。これら 2 つが組み合わさると、組み合わせ爆発が起こり、天文学的な数の化合物が生まれます。これにより、合成可能なすべての可能性を網羅するデータセットが生成され、探索範囲が広がります。
BioSolveIT テクノロジーは、膨大な数の分子コレクションの中から、重要でない分子にリソースを浪費することなく、創薬プロジェクトに関連する分子のみを取得します。それでも、ケミカルスペース全体の包括的な評価が実行され、結果としてすべての潜在的な分子が取り上げられる機会が確保されます。
ケミカルスペース — 化合物の商業的情報源
BioSolveITが開発した、infiniSee、infiniSee xREAL、Chemical Space Dockingは、超巨大なケミカルスペースから関連する化学情報を抽出するために利用可能です。取得した分子へのアクセスは、広く普及しているオンデマンドサービスと組み合わせることができます。当社のさまざまなパートナー企業のケミカルスペースから取得した化合物は、通常、数週間以内に購入し、お客様の元に配送することができます。
利用可能なケミカルスペース
infiniSeeとChemical Space Dockingでは以下のケミカルスペースから構造探索を行うことができます。
最新のケミカルスペースの情報は、開発元のウェブサイトをご覧ください。
table2:{ width:[20$20$20$20$20]$$
ケミカルスペース$化合物ベンダー$サイズ$購入可能※1)$合成可能$$
CHEMriya$OTAVA chemicals$1.2×1010$〇$ー$$
GalaXi$WuXi LabNetwork$1.2×1010$〇$ー$$
REAL Space$Enamine$7.0×1010$〇$ー$$
AMBrosia$AMBINTER$1.1×1011$〇$ー$$
Freedom Space$Chemspace$1.4×1011$〇$ー$$
eXplore$eMolecules$7.0×1012$〇$〇※2)$$
Knowledge Space$BioSolveIT$2.9×1014$ー$〇$$
}
※1: 化合物ベンダーにより合成され、数週間以内に入手可能。
※2: Cookbook (化学反応のリスト) を参照して、自社で合成可能。
BioSolveITでケミカルスペースを制覇
化合物の列挙には計算上の限界があるため、製薬業界では、社内のビルディングブロックと化学の専門知識を活用するために、コンビナトリアル ケミカルスペースを利用することが広く受け入れられています。結果へのアクセスのしやすさと知的財産の配慮が相まって、このアプローチの採用がさらに強化されています。これらのケミカルスペース内で新規化合物の探索範囲を拡大することは、計算上の制約による限界に対処するだけでなく、新たな発見の道を開き、製薬研究者がより幅広い潜在的な薬物候補を探索することを可能にします。
CoLibri

超大規模ケミカル スペースの心臓部は、採掘された化合物の合成アクセス性です。十分に簡単に合成できる化合物が、最終的に合成される化合物を決定します。
CoLibri は、合成のノウハウをケミカルスペースに変換し、膨大な数の仮想的でありながら合成アクセス可能な化合物を構成する、ケミカル スペース作成用のツールキットです。
一連のビルディングブロックと化学反応が与えられると、CoLibri は入力を処理し、*.space ファイルに書き込みます。これにより、社内のリソースが、創薬のための最大の探索場になります。
title:{コンポーネント}
コンポーネント
これらのコンポーネントは、BioSolveIT社の創薬プラットフォームを動かすギアとエンジンとして機能します。これらの計算ツールは、ドッキング (FlexX) やスコアリング (HYDE) など、創薬プロセスのさまざまな側面に対応するように設計されています。
コンポーネントは、既存のワークフローにシームレスに統合できるコマンドラインアプリケーションとしても利用できます。 このシームレスな統合により、当社のプラットフォームの適応性が向上し、研究者はこれらのツールを既存のプロセスにシームレスに組み込むことができるため、創薬の取り組みにおける効率と有効性が最適化されます。
幅広いパラメーターにより、柔軟に調整でき、プロジェクトの特定の要件に合わせて、より優れた正確な結果を得ることができます。
SeeSARに統合されたコンポーネント

HYDE
HYDEの結合評価は、脱溶媒和と相互作用という 2 つの主要な物理的駆動力に基づいて親和性を近似し、視覚化します。スタンドアロンのコマンドラインバージョンとしても利用できます。

FlexX
FlexX は、バーチャルスクリーニングに適した高速で柔軟なドッキング ソフトウェアです。ファーマコフォア拘束を適用し、化合物シリーズのドッキングとテンプレートドッキングを行えます。スタンドアロンのコマンドラインバージョンとしても利用できます。

FlexS
FlexS は、リガンドの配列を予測するためのツールです。指定されたリガンドのペアについて、FlexS は一方のリガンドが他方のリガンドに対してどのような立体配座と配向をとるかを予測します。スタンドアロンのコマンドラインバージョンとしても利用できます。

FastGrow
FastGrow は、3D構造に基づいてフラグメント伸張させるための、斬新で高速かつ正確な形状ベースのアルゴリズムを使用します。ユーザーは数秒以内に数万のフラグメントをスクリーニングし、結合部位を補完する最適化された候補構造を得ることができます。 スタンドアロンのコマンドラインバージョンとしても利用できます。
infiniSeeで実装されたコンポーネント

FTrees
Feature Trees (略称 FTrees) は、あいまいな類似性検索のための非常に効率的なツールで、特にバーチャルスクリーニングで明白でないヒットの発見に適しています。スタンドアロンのコマンドラインバージョンとしても利用できます。

SpaceLight
SpaceLight は、分子フィンガープリントの類似性に基づいて、クエリ化合物の類似化合物を広大なケミカルスペースからスクリーニングします。スタンドアロンのコマンドライン バージョンとしても利用できます。

SpaceMACS
SpaceMACS は、部分構造の類似性に基づいて、コンビナトリアルケミカルスペースから分子を抽出します。得られた結果には、関心のある分子モチーフが含まれます。スタンドアロンのコマンドラインバージョンとして利用できます
その他のコンポーネント

CoLibri
CoLibri は、ケミカルスペース探索のためのツールキットで、合成知識をケミカルスペースに変換し、バーチャルでありながら合成可能な膨大な数の化合物を構成します。スタンドアロンのコマンドラインバージョンとして利用できます。

Conformator
Conformator は、分子構造に関して非常に高い精度で、指定された分子セットの 3D 分子アンサンブルを生成します。配座の多様性に重点が置かれています。スタンドアロンのコマンドラインバージョンとして利用できます。
title:{動作環境}
動作環境
BioSolveIT社創薬支援ツールは以下のOSに対応しています。詳細はお問い合わせください。
table1:{
width:[25$50]$$
ツール名$対応OS$$
SeeSAR$Windows, macOS, Linux$$
infiniSee$Windows, macOS, Linux$$
infiniSee xREAL$Windows, macOS, Linux$$
HPSee$Windows, Linux$$
HYDE$Windows, macOS, Linux$$
FlexX$Windows, macOS, Linux$$
FlexS$Windows, macOS, Linux$$
FastGrow$Windows, macOS, Linux$$
Conformator$Windows, macOS, Linux$$
FTrees$Windows, macOS, Linux$$
SpaceLight$Windows, macOS, Linux$$
SpaceMACS$Windows, macOS, Linux$$
CoLibri$Windows, Linux$$
}
}:tab