


Direct Force Field
(高精度シミュレーション支援ソフトウエア)
tab:{
title:{概要}
Direct Force Fieldとは?
DFF「力場パラメータが足りなくて計算できない」 「化学的に誤った構造を結果として与える」 これらは、分子シミュレーションソフトウェアに付属する力場パラメータを利用して計算を行うと、しばしば直面する問題です。 このような問題は、力場パラメータを自作するか、論文からデータを探し出してそれを利用することでしか、解決できません。 パラメータを自作するにはノウハウが必要で一部の専門家にしかできません。また、論文のパラメータデータの精度は、目的が同じでない限り、保証されません。 Direct Force Fieldは、「さまざまな分子シミュレーションソフトウェアに高精度な力場パラメータを提供すること」を目的に開発され、このような力場パラメータの問題を解決します。
パンフレット

Direct Force Field パンフレットPDF版(0.4 MB)
Aeon Technology社 ウェブサイト
Direct Force Fieldの開発元であるAeon Technology社ウェブサイトもあわせてご覧ください。

title:{特長}
特長
最も高い精度を持つ力場パラメータ
Direct Force Fieldでは、独自の手法によりパラメータの代用の問題を解決し、分子系に最適な力場パラメータをアサインすることが可能です。また、登録されている力場パラメータは、開発者のCOMPASS*1等の力場開発における経験と知識に基づいて、作成、検証されています。そのため、Direct Force Fieldでは現在考えられる最も高い精度を持つ力場パラメータが提供されています。
他の分子シミュレーションソフトウェアで直接利用できるファイル
力場データベース*2による目的分子への力場パラメータのアサインはコマンド1つで自動的に行われ、サポートしている分子シミュレーションソフトウェアに対応したフォーマットで、直ちに保存することができます。力場パラメータの情報はすべてテキストファイルで出力されますので、必要に応じて変換していただくことでサポート外の分子シミュレーションソフトでもご利用いただくことが可能です。
用途に応じた力場データベース
材料設計向けまたはライフサイエンス向けの力場DBいずれかが付属します。いずれも有機低分子、合成高分子、薬物分子等の有機分子全般に対応できるようパラメータを作成しています。材料設計向け力場DBは関数形にAeon社オリジナルのTEAMを採用し、ライフサイエンス向け力場DBには関数形にAMBERを採用しています。また、特定の目的に対して利用できるよう、材料設計向けにはゼオライトに対応した力場、ライフサイエンス向けにはAMBER95を基にした力場が追加されています。
力場パラメータの作成
量子化学計算の結果から力場パラメータの作成が可能です。非常に複雑なポテンシャル面に、力場のポテンシャル関数を効率よくフィットするパラメータ決定手法を備えており、量子化学計算のデータから信頼性の高い力場パラメータを高速に開発できます。 一般の力場パラメータのデータは原子タイプ割付ルールとパラメータデータが一組となっています。これ対し、Direct Force Fieldでは複数組のデータを同時に取り扱えるようにするため、そのデータはデータベースの形式で管理されています。このデータベースを「力場データベース」と呼びます。
*1. Sun, H. J. Phys. Chem. B, 1998, 102, 7338-7364
*2. 一般の力場パラメータのデータは原子タイプ割付ルールとパラメータデータが一組となっています。これ対し、Direct Force Fieldでは複数組のデータを同時に取り扱えるようにするため、そのデータはデータベースの形式で管理されています。このデータベースを「力場データベース」と呼びます。
title:{パッケージ}
パッケージ
Direct Force Fieldでは、各機能をモジュールに分割しており、目的に応じてご利用いただけるようパッケージ化して提供しています。パッケージはStandard、Professionalの2種類が用意されています。いずれのパッケージにもAeon社オリジナルの力場DB、材料設計向けTEAM_Family(関数形TEAM)、生命科学向けAMBER_Family(関数形AMBER)の内の1つが付属します。
Standard
LAMMPS、AMBER、GROMACSなどの分子動力学計算ソフトウェアで、最高精度の力場パラメータを、手軽に利用したい方のための製品です。既存の分子動力学計算ソフトウェアが提供するパラメータセットでは十分な精度が得られない場合に有用です。
Professional
広範な分子をカバーしているAeon社提供の力場データベースでも、目的分子によっては力場パラメータが不足して計算が実行できない場合やパラメータの精度をより高くしたい場合があります。Professionalには、新たに力場パラメータを作成して、データベースに登録する機能があるので、このような場合に有用です。予め用意されたテンプレートの関数項を自由に組み合わせた、ユーザオリジナルの力場タイプの定義も可能です*3。一般的な力場タイプ同様、ユーザ定義の力場タイプに対しても同じようなパラメータ作成法が利用できますので、力場ポテンシャル関数の開発に役立てることができます。

*3. ユーザオリジナルの力場に対応した力場データベースは構築できません。 ユーザオリジナルの力場を利用する場合は、目的分子ごとにパラメータセットを作成し、1つのファイルとして保存することとなります。
title:{モジュール}
モジュール
パッケージを構成する4つのモジュール機能について説明します。
力場データベース
(TEAM_Family、AMBER_Family)

一般的な力場では、原子タイプ割付ルールと力場パラメータは一組のみ取り扱われますが、力場データベース(以下力場DB)では、異なる原子タイプ割付ルールと力場パラメータをもつ力場を、複数同時に整合性を保ちながら統合し、利用する機能を備えています。この機能により、力場タイプが同一であれば、一部をユーザオリジナルの力場、他の部分をDFFの提供する力場というようにして、別々に開発された力場パラメータを矛盾なく1つのパラメータセットとして利用することが可能です。また、原子タイプ割付ルールを簡単に編集する機能を備えているので、デフォルトより詳細なユーザ独自の割付ルールを作成することも可能です*4。これらにより、力場パラメータ代用の問題と原子タイプの重複による矛盾を解決しながら、力場パラメータの精度と拡張性を同時に向上させています。
製品付属する力場DBには、TEAM_Family、AMBER_Familyの2つが用意されており、用途に応じていずれかの力場DBを選択していただくことになります。材料設計向け力場DB、TEAM_Familyは関数形にAeon社オリジナルのTEAMを採用し、有機低分子、合成高分子、イオン液体、ゼオライト用のパラメータが登録されています。ライフサイエンス向け力場DB、AMBER_Familyは関数形にAMBERを採用し、TEAM_Family同様、有機低分子、合成高分子、イオン液体のパラメータが登録されており、さらに公開されているAMBER95 のパラメータが登録されています。
*4. Professionalのみの機能です。
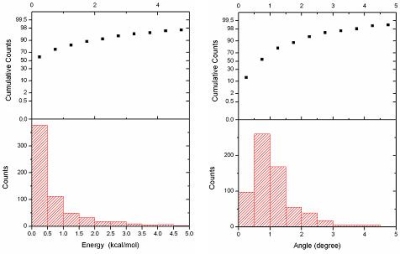
- 力場データベースにおけるデータフィッティングの信頼性
量子化学計算とのコンフォメーションのエネルギー誤差(左)と最適化構造の結合角の誤差(右)の分布のグラフとその積算率。コンフォメーションのエネルギー誤差が3kcal/mol以下のものが95%、結合角の誤差は3度以下のものは約98%と、サンプリングに利用した量子化学計算と同程度の精度が望めます。
- 力場パラメータ代用の問題
力場の精度は、力場パラメータの数値の精度だけでなく、分子中の多様な原子の環境を解釈して如何に妥当な力場パラメータを原子にアサインできるかに大きく依存します。一般の力場の二面角(A-B-C-D)の力場パラメータは、連なる4原子の2番目(B)と3番目(C)の原子タイプによりアサインされます。このような場合、1番目(A)と4番目(D)の原子がどのような原子タイプであっても、全く同じ力場パラメータを利用してしまうため、二面角の回転障壁の形が全く異なってしまうことが多々あります。このようなことを力場パラメータの代用の問題といいます。
GUI機能
分子構造の構築、可視化、さまざまな分子フォーマットの読み込みを可能にします。さらに、力場の開発だけでなく、一般的な分子モデリングのツールとしても利用できます。低分子液体モデリング機能、自動電荷グループアサイン機能等が搭載されています。
シミュレーション機能
DFFの力場データベースで得られた力場パラメータや開発された力場パラメータを、DFFの分子シミュレーションエンジンやサードパーティの分子シミュレーションエンジンで利用するための機能です。DFFの分子シミュレーションエンジンは力場パラメータの検証やVDWパラメータ作成に必要な分子動力学計算データを用意するのに利用されます。サードパーティの分子シミュレーションエンジンに対しては座標データ、トポロジーデータ、原子タイプデータ等、計算に必要なデータの作成を行います。
力場パラメータ開発機能

量子化学計算の結果から力場パラメータの作成するための機能です。非常に複雑なポテンシャル面に、力場のポテンシャル関数を効率よくフィットするパラメータ決定手法を備えており、量子化学計算のデータから信頼性の高い力場パラメータを高速に開発できます*5。 本機能には、力場パラメータを作成するのに必要な部分構造(フラグメント)に対して、量子化学計算データを作成、読み込みをするためのインターフェースが含まれています*6。
力場パラメータ作成には、フラグメントに対して、最安定構造の探索、基準振動解析、パラメータフィッティングに利用する配座の一次微分の計算を行います。本来、これら手順は、全てユーザが一つ一つコマンドを実行して行う手間のかかる作業ですが、1つのボタンをクリックするだけで自動的に行うこともできます*7。
凝集系のシミュレーションで高い精度を要求されるvan der Waals (VDW)パラメータについては、系の密度と蒸発熱の実験値から自動的にVDWパラメータをフィッティングする独自の手法が搭載されています*8。 さらに、予め用意されている力場のポテンシャル関数でよく利用されるさまざまな項のテンプレートを自由に組み合わせることで、ユーザオリジナルの関数による力場パラメータ開発も可能です。これにより、ライフサイエンスからマテリアルサイエンスまで、さまざまな分子シミュレーションに対応できる力場を開発することが可能です。
*5. Sato, F.; Hojo, S.; Sun, H. J. Phys. Chem. A, 2003, 107, 248-257
*6. Gaussianに対応。量子化学計算ソフトウェアGaussianが別途必要です。
*7. GaussianがDFFの起動できるPCにインストールされている必要があります。
*8. Sun, H. Fluid Phase Equilibria, 2004, 217, 59-76.
title:{適用事例}
適用事例
DFFとSciMAPSによる二次電池用電解質の検討
有機電解質を利用したリチウムイオン二次電池は携帯電話、パソコンから電気自動車までさまざまな用途で実用化されています。電池を構成する有機液体電解質については、様々な化合物を組み合わせることで、軽量化、高安全性、高耐久性などを実現する電解質材料の開発が重要となっています。本稿では、電解質によく利用される有機低分子とLiTFSAの塩からなる系を構築し、密度、拡散定数を予測し、実験値と比較しました。また、動径分布関数より配位分布関数を求め配位状態の比較を行いました。詳細は下記pdfをご参照ください。
TEAM力場による熱伝導率計算
電子機器の小型化・高機能化により増大する発熱方向の制御、車載電子機器の動作温度の維持、排熱再利用による未利用エネルギーの削減などにおいて、材料による伝導熱の制御(サーマルマネージメント)は1つの課題となっています。原子の分布、官能基の配向等の原子・分子レベルでの挙動と熱伝導率の関係を詳細に調べられる分子シミュレーションによるアプローチは、ナノスケールの情報によるサーマルマネージメント材料の探索手法として利用されています。本稿ではとDirect Force Fieldの力場TEAMと材料設計支援プラットフォームSciMAPSの分子構築機能、熱伝導率計算機能を用いて、低分子、高分子、アモルファスと結晶の3つの観点から検討した事例を紹介します。詳細は下記pdfをご参照ください。
メタノール用力場の作成
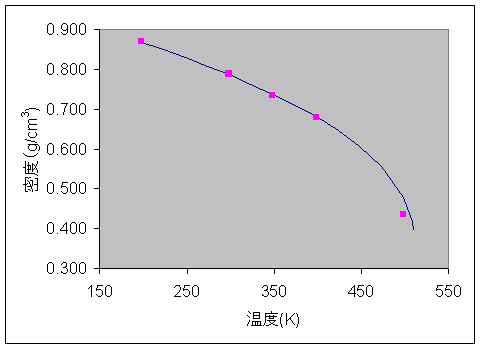
Direct Force Field では、密度や蒸発熱を利用してVDWパラメータを自動作成する機能により、凝集系シミュレーションでも精度の高い計算ができるパラメータを手軽に作成できます。この機能は、 専門家がノウハウを使って数週間かかっていたVDWパラメータ作成を自動的に1、2日程度で作成できるようになり、 凝集系に対応したパラメータ作成をより身近なものにします。Valenceパラメータと電荷パラメータのフィッティングにはB3LYP/6-311G**の量子化学計算結果を用い、VDWパラメータのフィッティングには常温常圧下(温度: 298 K、圧力: 0.1 MPa)でのメタノールの密度と蒸発熱を用いました。 作成した力場パラメータは、分子構造、内部回転エネルギー障壁や基準振動はもちろんのこと、液体メタノールの平衡状態を精度良く予測しました。定温定圧(NPT)の分子動力学シミュレーションにより、液体メタノールの等圧膨張曲線を描くと下図のようになり、 Direct Force Field により自動的に得られたパラメータが密度の温度依存性を精度良く再現していることがわかります。
title:{論文リスト}
論文リスト
DFFに関連する学術論文です。
-
Gong, Z. and Sun, H
Pressure-viscosity relation of 2,2,4-trimethylhexane predicted using all-atom TEAM force field
Fluid Phase Equilibria, 2019, 497 ,64-70
-
Gong, Z. and Sun, H
Extension of TEAM Force-Field Database to Ionic Liquids
J. Chem. Eng. Data, 2019, 64, 9, 3718–3730 -
Huang, H; Wu, L; Xiong, H; Sun, H
A Transferrable Coarse-Grained Force Field for Simulations of Polyethers and Polyether Blends
Macromolecules, 2019, 52, 1, 249–261 -
Gong, Z; Wu, Y; Wu, L; Sun, H
Predicting Thermodynamic Properties of Alkanes by High-throughput Force Field Simulation and Machine Learning
J. Chem. Eng. Data, 2019, 64, 9, 3718–3730 -
Gong, Z; Sun, H; Eichinger, B. E.
Temperature Transferability of Force Field Parameters for Dispersion Interactions
J. Chem. Theory Comput., 2018, 14, 7, 3595–3602 -
Huang, H; Cao, F; Wu, L; Sun, H
All-Atom and Coarse-Grained Force Fields for Polydimethylsiloxane
Mol. Simul.,2017, 43, 1513–1522. -
Wu, L; Chen, L; Sun, H
On accuracy of predicting densities and solubility parameters of polymers using atomistic simulations
Mol. Simul.,2017, 43, 510-518.
-
Sun, H; Jin, Z; Yang, C; Akkermans, R. L. C; Robertson, S. H; Spenley, N. A; Miller, S; Todd, S. M
COMPASS II: Extended Coverage for Polymer and Drug-like Molecule Databases
J. Mol. Model., 2016, 22, 47. -
Jin, Z; Yang, C; Cao, F; Li, F; Jing, Z; Chen, L; Shen, Z; Xin, L; Tong, S; Sun, H
Hierarchical atom type definitions and extensible all-atom force fields
J. Comput. Chem., 2016, 37, 653−664. -
Sun, H
COMPASS: An ab Initio Force-Field Optimized for Condensed-Phase Applications Overview with Details on Alkane and Benzene Compounds
J. Phys. Chem. B,1 998, 102 (38), 7338-7364. -
Sun, H.
Automatic Parameterization of van der Waals Forces-With Application on Prediction of Fluid Densities
First Industrial Fluid Properties Simulation Challenge, 2002 Annual Meeting, AIChE. -
Sato, F.; Hojo, S.; Sun, H.
On the Transferability of Force Field Parameters-With an ab Initio Force Field Developed For Sulfonamides
J. Phys. Chem. A , 2003, 107,248-257
title:{動作環境}
動作環境
下記オペレーティングシステムに対応しています。
- Windows 10、11
- Linux
}:tab