


MOE - Structure-Based Design
tab:{
title:{概要}
Structure-Based Design は、標的タンパク質や受容体の立体構造情報に基づいた分子設計法です。MOEの Structure-Based Design に関連する機能として、分子表面の特性解析や化合物のドッキングシミュレーション、結合部位での対話的な分子設計、フラグメントを用いた分子設計によって新規リガンド候補を探索・設計します。また、タンパク質-リガンド相互作用解析機能を組み合わせて、妥当な候補構造の絞り込みや重要な相互作用の検出が可能です。
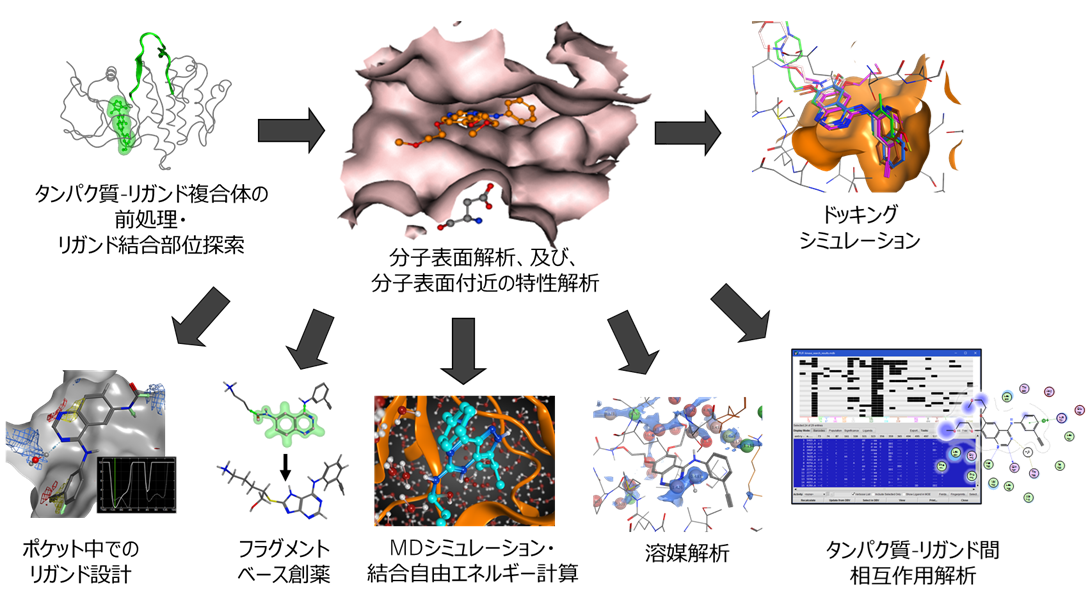
title:{新規リガンド候補の探索と設計}
ドッキングシミュレーション
標的タンパク質の活性部位中に、化合物を効率的に配置し、その安定な結合配座を予測します。評価関数には、静電、ファンデルワールス、溶媒和の各相互作用エネルギーと、接触表面積に基づいた関数、電子密度マップとの重なりに基づいた関数などから選択し、バーチャルスクリーニングが行えます。タンパク質構造を可動にしたInduced-Fitドッキングにも対応しています。以下のようなドッキング手法を利用できます。
- 標準ドッキング
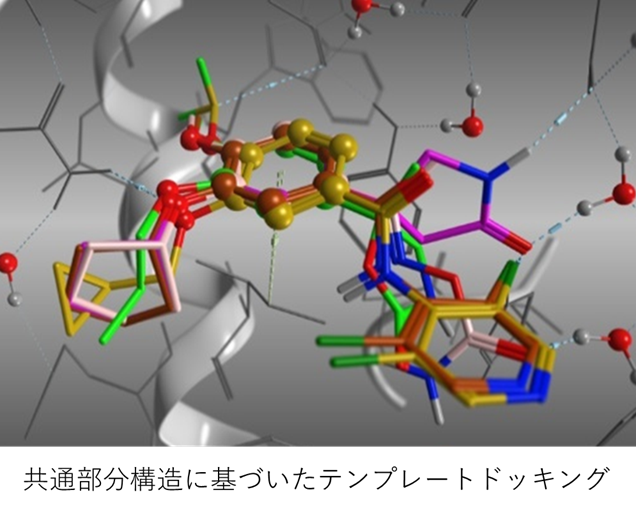
- テンプレートドッキング
- Covalent Docking
- 電子密度フィッティング
- タンパク質(ペプチド、核酸)-タンパク質ドッキング
- 外部プログラムとの連携(FlexX、GOLD、SurflexDock)
- ASEDock: カスタムプログラム
結合部位での分子設計
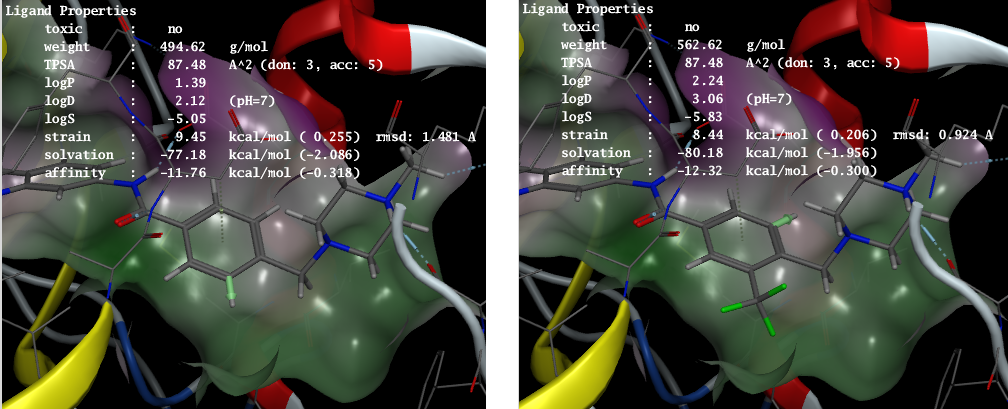
受容体に結合したリガンド構造を、より良好な活性や物性をもつように対話的に編集できます。結合したリガンド構造のうち、置換基を付加可能な領域は分子グラフィックスの緑のベクトルとして表示されます。リガンド構造の分子量やTPSA、logP、logD、logSなどの物性、リガンドのひずみエネルギー、受容体との親和性、毒性の有無などの値を確認しながら、リガンドの改変が可能です。フラグメントライブラリーからの置換基の自動探索や、2Dスケッチャーを用いた分子構造の改変も可能です。
母核構造置換/フラグメント付加・連結
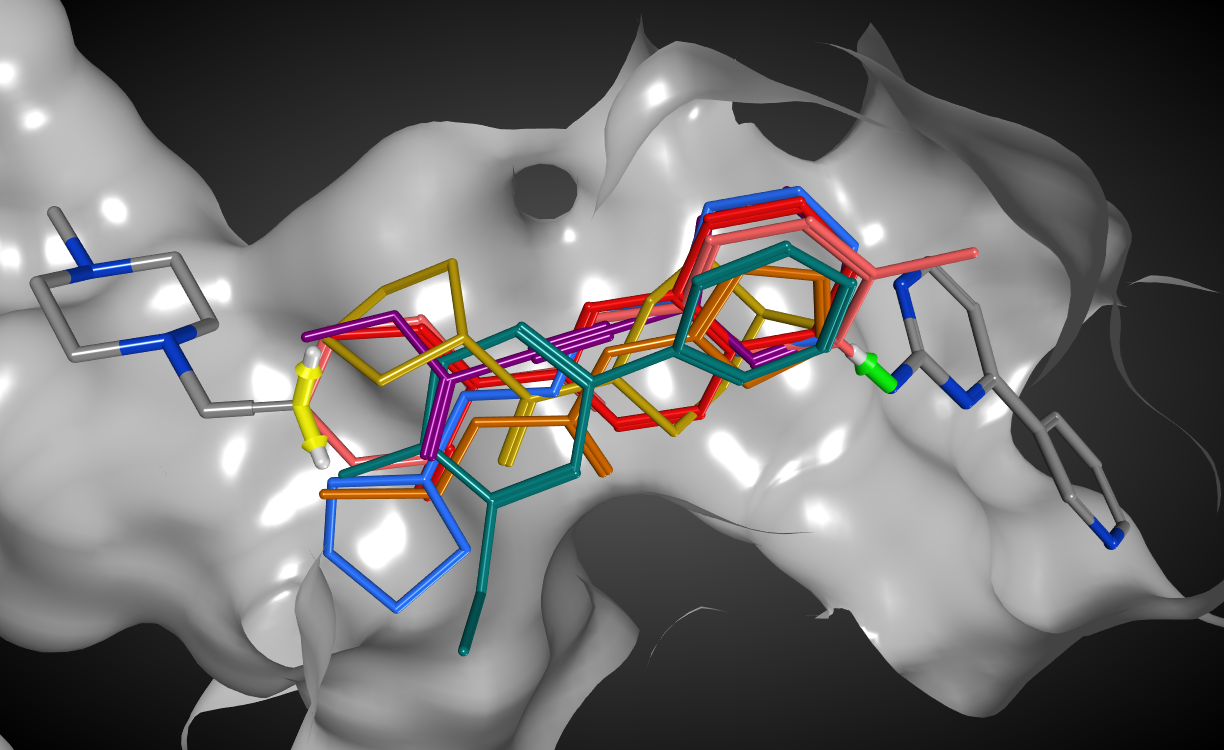
結合部位中のリガンド構造に対して、母核構造の置換や官能基の付加、複数フラグメントの連結を行うことで、新規リガンド候補を提案します。MOEに付属する150万件以上のフラグメントデータベースもしくはin-houseデータベースからポケットに適合するフラグメントを高速に検索します。環化や環縮合を伴う結合条件も指定できます。
得られた構造は、分子記述子、QSARモデル、類似度、ファーマコフォアモデルによって絞り込むことができます。さらに、合成可能性スコアやドッキングスコアによる順位付けを行うことで、実験に使用する構造の優先度を決定できます。
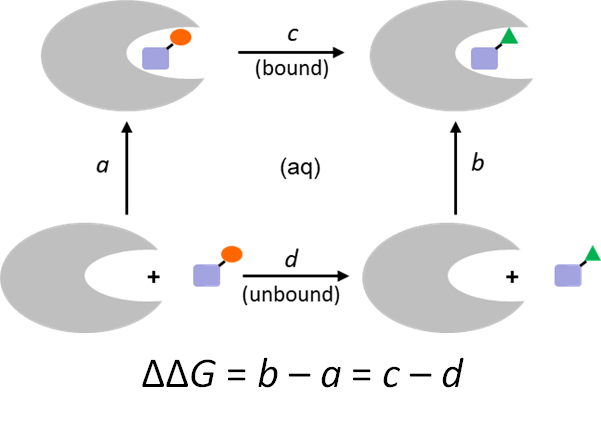
結合自由エネルギーの推算
Amber*の熱力学的積分法(Thermodynamic integration, TI)のインターフェースにより、標的タンパク質と複数のリガンド間の相対的結合自由エネルギーΔΔGを簡単に計算できます。化合物を順位付けすることで、合成する化合物数を絞り創薬研究をより効率化できます。
* 別途Amberプログラムが必要
title:{タンパク質-リガンド間相互作用解析}
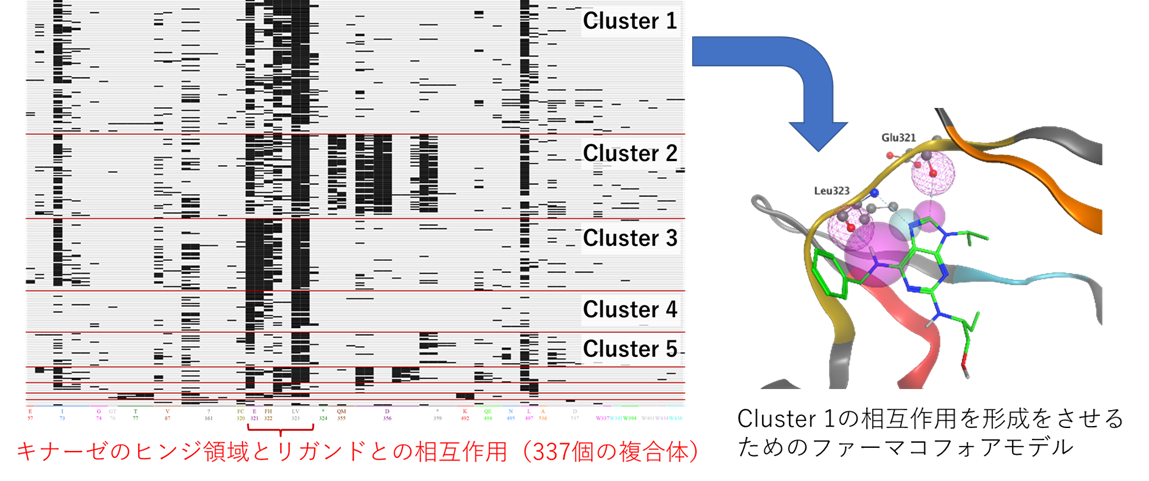
Protein Ligand Interaction Fingerprints (PLIF)
タンパク質-リガンド間の相互作用の種類と強さを固定長のフィンガープリントで表現し、複数の結合状態を統計的に解析します。ドッキングの結果や複数の複合体構造に含まれる相互作用を解析することで、共通する相互作用の検出、活性/不活性に関連する相互作用の検出、活性/不活性を分類する相互作用組み合わせルールの抽出、相互作用によるクラスタリング、相互作用を満足するためのファーマコフォアの導出などが行えます。既存のリガンドとの相互作用の類似度も計算できます。
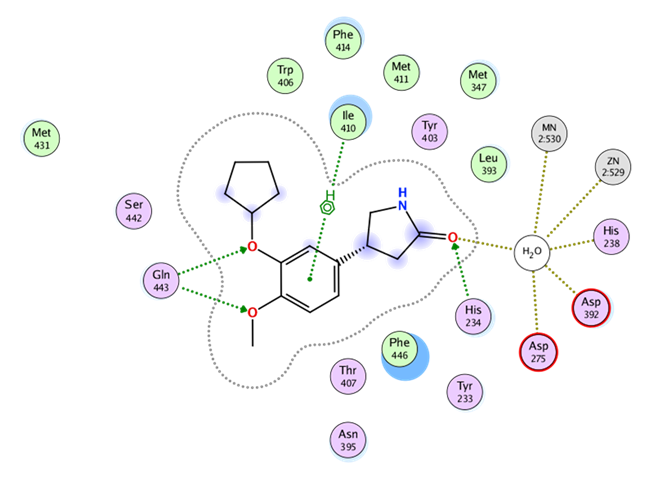
リガンド結合部位の2次元図
タンパク質-リガンド複合体からリガンド結合部位の2次元図を表示します。周辺残基の位置関係や相互作用、溶媒露出度が直感的に分かるように表示されます。
水素結合、イオン結合、π-π相互作用、CH-π相互作用、カチオン-π相互作用、ハロゲン結合、配位結合などの相互作用を、それぞれ色分けして表示します。
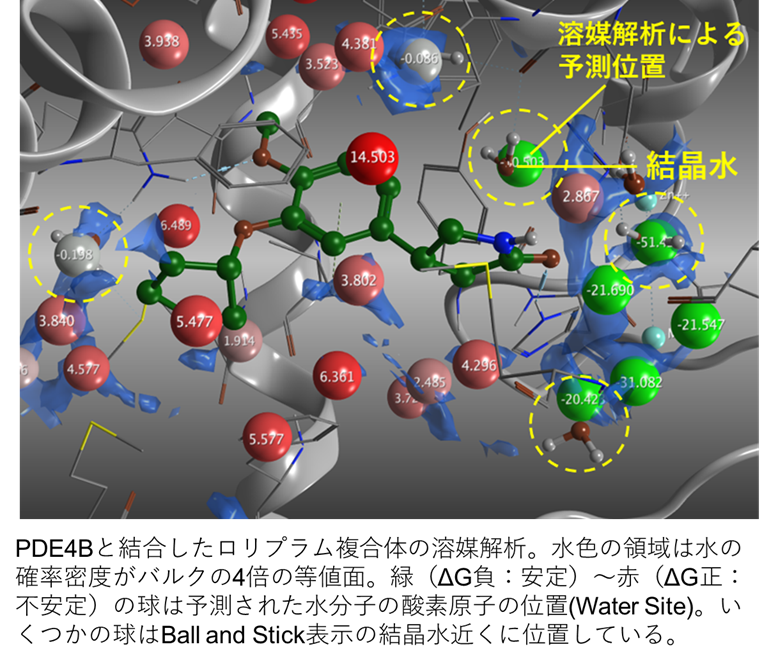
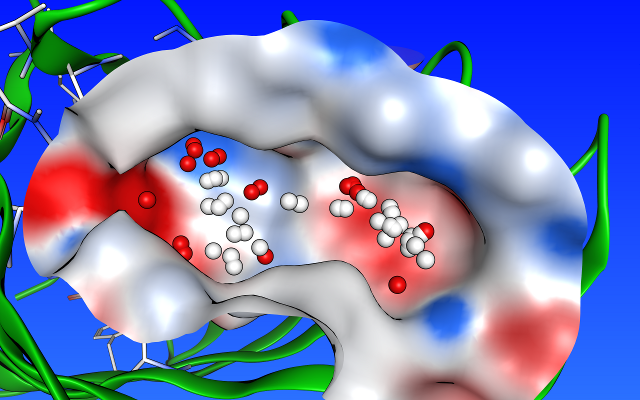
溶媒解析
3D-RISM法により、高速かつ高精度に水分子の密度分布や溶媒和自由エネルギーを求め、タンパク質やリガンドの周辺に存在する水を可視化します。水素結合の評価、水による相互作用ブリッジの理解、疎水性相互作用など、相互作用に関わる溶媒効果を詳細に検討できます。
また、溶媒解析の結果を基にして、水分子の位置(Water Site)とその溶媒和自由エネルギーも予測します。Water Siteは、タンパク質-リガンド相互作用における重要な水分子や、疎水性原子への置き換えなど、リガンド設計における情報を提供します。
MOE Solvent Analysis
分子動力学計算(MDシミュレーション)
MD(Molecular Dynamics)シミュレーションは、分子の動的な挙動を解析できます。例えば、リガンド-受容体間の相互作用解析や、タンパク質の構造変化、リガンドやペプチドの配座解析、水分子の役割、イオンや添加剤の影響、結合自由エネルギー計算など創薬プロセスにおける多くの情報を提供します。
MOEには、Amber、NAMD*の外部MDツールのインターフェースが搭載されており、ツールに必要な入力ファイルは全てMOEが自動で作成できます。結果の取り込みと解析もMOE上で可能です。
*別途Amber、NAMDプログラムが必要
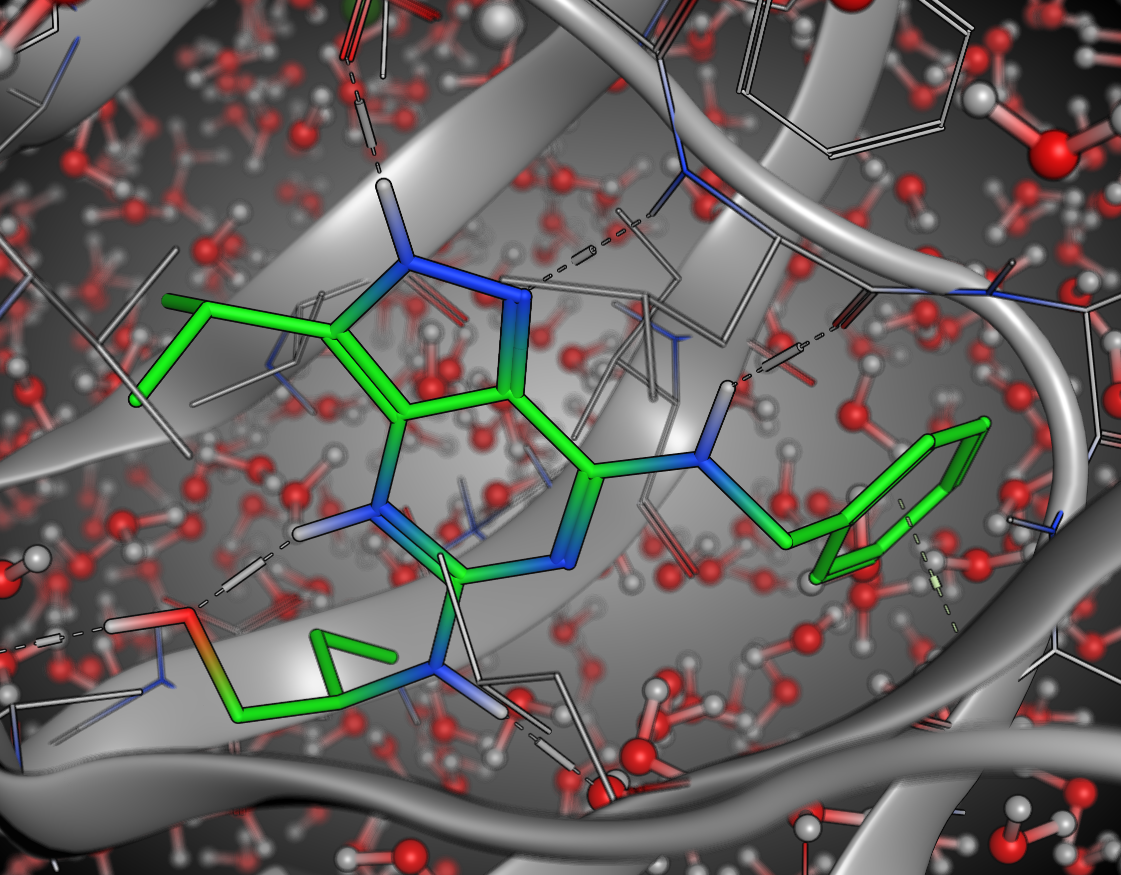
title:{分子構造の前処理と分子表面解析}
複合体構造の前処理
相互作用解析やドッキングシミュレーション等を行う前の、受容体構造やリガンド構造の適切な前処理は重要なステップで、その結果の精度に大きく影響します。MOEは、分子構造における適切な前処理機能が搭載されています。標的タンパク質の主鎖や側鎖構造の補完、相互作用に関与しない水分子の除去、Protonate3Dによる水素原子付加状態の最適化、リガンドとその結合部位の構造最適化などの前処理を一括で行うことが可能です。処理内容の編集や保存も可能です。
分子表面解析
分子の表面形状として、溶媒接触表面とInteraction表面の2種類を描画することができます。溶媒接触表面は、水分子との接触可能な表面を描画し分子の大きさや形状を確認できます。Interaction表面は、受容体原子とプローブ球とのvan der Waals相互作用エネルギーが0 kcal/molとなる面を描画し原子間接触を判定できます。それぞれの表面は、水素結合性、静電ポテンシャル、疎水性-親水性、凹凸などを指標にした色分けにより、分子表面の特性を視覚的に解析できます。
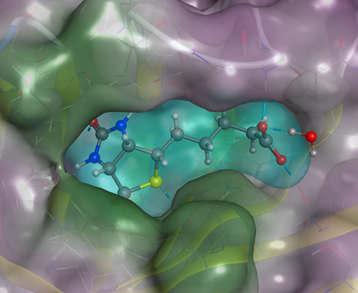
結合部位中の特性解析
表面に対する相互作用相手の原子として好まれる特性領域をグラフィカルに表示します。領域は静電ポテンシャルや、PDBデータを元に解析された相互作用原子ペアの統計解析、相互作用エネルギーなどを基に予測されます。複合体構造における相互作用の安定性に寄与している部分構造の予測や、分子設計の改変に適した箇所を検出できます。
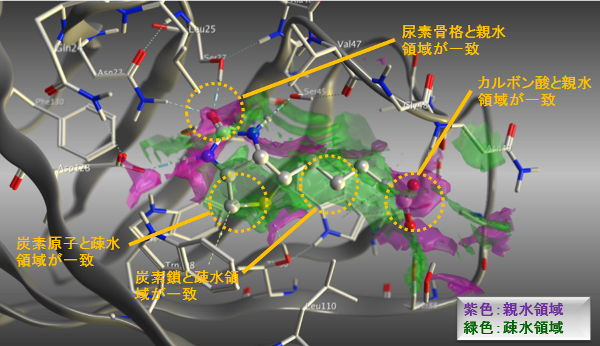
リガンド結合部位探索
タンパク質の活性部位候補となりうるポケット構造を自動的に検出することができます。各ポケットに対して、アルファ球と呼ばれる疎水性(白色)または親水性(赤色)の小球を配置し、各ポケット内における疎水性・親水性領域の分布をグラフィカルに表示します。検出した結合部位候補は、PLB indexスコアで順位付けされより有望なDruggableなサイトを選べます。
この探索機能は、ドッキングシミュレーションのためのリガンド結合部位の選定だけでなく、既存のリガンドの修飾や新規リガンドの設計、ファーマコフォアモデルの作成にも利用することができます。
}:tab