


MOE - バーチャルスクリーニング
tab:{
title:{概要}
MOEはバーチャルスクリーニングにおける各段階において多くの有用な機能を提供します。例えば、化合物ライブラリーの作成では、化学反応ルールに基づいて試薬データベースから網羅的に化合物群を生成できるコンビナトリアルライブラリー設計の機能があります。化合物の前処理では、分子構造の3次元化や指定したpHにおける妥当なイオン化状態への変換を行えるWashや高速な配座生成ができるConformation Importと呼ばれる機能があります。化合物フィルタリングでは、記述子、部分構造、フィンガープリント、QSAR/QSPR、ファーマコフォア検索、ドッキングなどの様々な機能が使用できます。MOEではそれらの機能を組み合わせて膨大な化合物ライブラリーから目的の化合物群を効率よく抽出できます。

title:{ライブラリー}
化合物ライブラリーの提供
リードライク化合物データベース
MOEは、リードライクフィルターを満足する約65万化合物を含むデータベースを提供します。これらの化合物群は、試薬供給会社の公開カタログから収集されたものです。このデータベースは配座解析済み(約3,470万配座)であるため、すぐにファーマコフォア検索やドッキングシミュレーションなどに使用できます。
日本国内の試薬供給会社が提供するMOE版化合物カタログ
試薬供給会社と提携して、MOE版化合物カタログを発行しています。詳細は化合物カタログのページをご参照ください。
化合物ライブラリーの設計
コンビナトリアルライブラリー設計
化学反応ルールに基づいて試薬の組み合わせから網羅的な化合物ライブラリーの構築とスクリーニングができます。60以上の化学反応ルールと試薬ライブラリーが提供されています。また、スケッチャーを用いて独自の化学反応ルールを定義できます。
逆合成解析に基づくライブラリー設計
化合物群を、逆合成解析により得られたRECAPルールに基づき、結合を切断してフラグメント化します。また、フラグメントを相補的な組み合わせ の反応点で結合させることで、合成経路の設計が容易な新規化合物を構築します。市販HTS化合物などから独自に収集した約12万件のフラグメ ントを登録したRECAPフラグメントデータベースも提供しています。

多様性バーチャルライブラリー設計
指定した分子数で多様性をもつライブラリーを構築できます。最初に指定した数の分子を構築した後、多様性が十分に大きくなるまで新しい分子を構築してライブラリーの更新を続けます。この手法により、冗長性の低い、多様性をもった任意の大きさのライブラリーを設計することが可能です。活性化合物を指向したライブラリー設計のために、分子量やlogP値での制限や確率モデルを使用したフィルタリングも行えます。
ライブラリー補間
複数のライブラリーから特性の重複しない化合物を組み合わせ、多様性の大きいライブラリーを作成します。既存のライブラリーに新しい化合物データを追加する際に効率良く化合物を選抜して組み合わせられます。
化合物ライブラリーの前処理
・分子構造の標準化(Wash)
公共のデータベースや化学構造式描画ソフトウェアから取得した分子データはシミュレーションに適さないことがあります。例えば、分子が二次元構造、つまり構造式であったり、不適切なイオン化状態であったり、解析に不要な塩、溶媒分子、イオンを含むことがあります。したがって、これらの分子データを使用してさまざまな解析を始める前に標準化しておくことは重要です。Washはこれらの問題を解決できる分子の標準化の機能です。ナトリウム、カリウムなどのアルカリ金属との結合を切断し、メインとなる分子以外の溶媒分子やイオンなどを削除できます。また、pHに基づくイオン化状態の予測と水素付加、分子構造の3次元化ができます。
・高速な配座生成(Conformation Import)
バーチャルスクリーニングにおいて、ドッキングシミュレーションやファーマコフォア検索を行う場合、分子の三次元構造(配座)が必要です。大量の化合物の配座生成に適した機能として、Conformation Importがあります。この機能では、化合物をフラグメントに分割し、各フラグメントについて配座解析を行い、 それらのフラグメント配座を結合することで化合物の安定配座を高速に発生させます。
title:{記述子/部分構造}
化合物ライブラリーには、望まない物性(溶解度が低い、毒性がある、ドラッグライクでない、など)を持つ化合物が含まれている可能性があります。以下の記述子や部分構造を用いたフィルタリングでこれらの化合物を取り除くことで、より有望な化合物群に絞り込み、次ステップのより高度なスクリーニングを効率化できます。
記述子によるフィルタリング
分子記述子(記述子)は、分子の物理化学的な特性を数字で表現したものです。MOEには800種類以上の記述子が標準で搭載されており、さらに無償で提供されるアドオンプログラムを追加すれば、その数は数千種類以上になります。一般的な記述子、分子量、logP(オクタノール/水分配係数の対数)、logS(水溶解度の対数)、水素結合ドナー/アクセプター原子の数、TPSA(極性表面積)などがあります。バーチャルスクリーニングでは、これらの記述子を大量の化合物について一括計算し、その値に閾値を設けて化合物群を絞り込めます。バーチャルスクリーニングで有用な記述子としては、上記に加えて、合成可能性、反応性、変異原性、ドラッグライク、リードライク記述子などがあります。さらに、ユーザーはMOEの開発環境を使用して、独自の記述子を簡単に追加できます。

部分構造によるフィルタリング
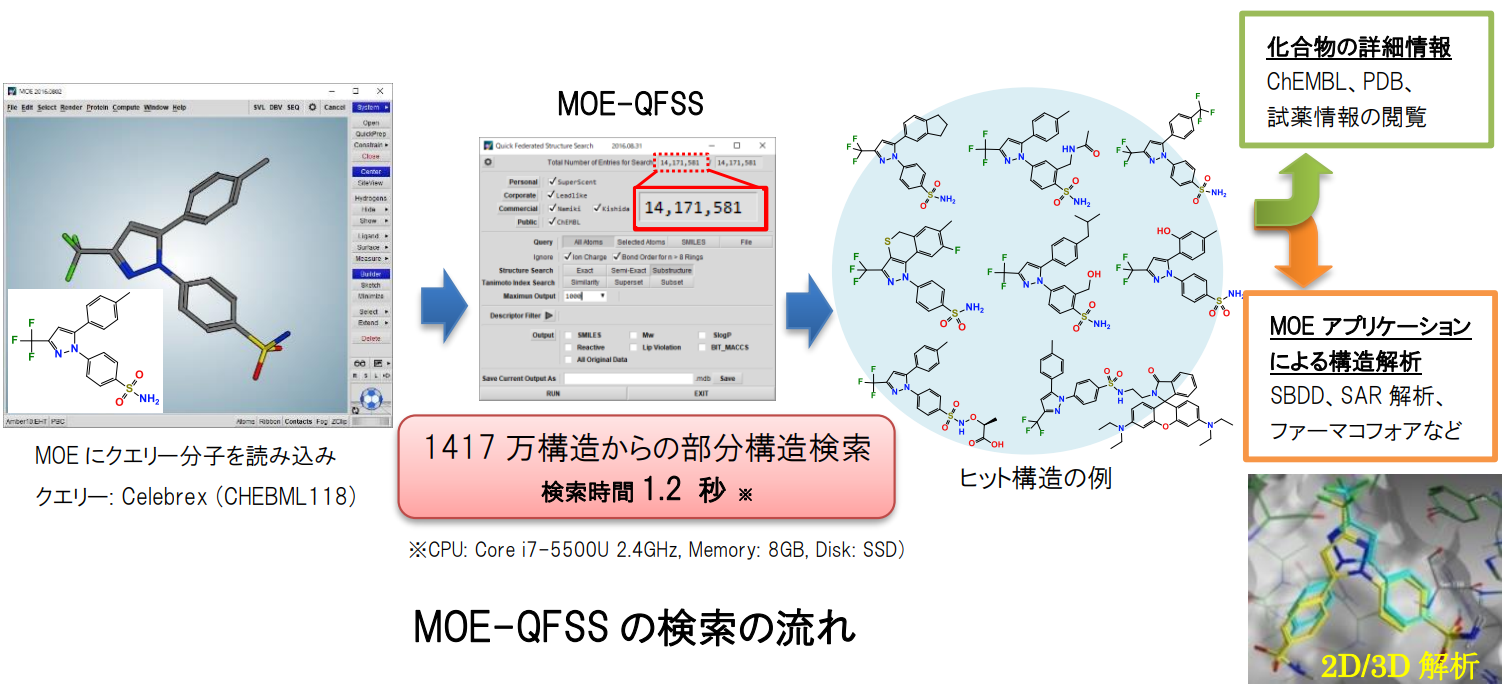
部分構造によるフィルタリングでは、SMILES/SMARTS表記を用いた構造検索が利用できます。SMILES/SMARTSは化学構造式を文字列として記述したもので多様な分子構造が表現できます。また、当社が開発したMOEのアドオンプログラムのMOE-QFSS(Quick Federated Structure Search)を使用すれば、複数のMDBに保存されている数千万件の化合物構造データに対して、数秒以内で部分構造検索や類似構造検索が行えます。データを効率的に管理することでメモリー消費を押さえているため、一般的なラップトップPCでも超高速な構造検索を実現しています。ユーザーは、MOE-QFSSを用いて、試薬カタログ、公共データベースなどから目的化合物を高速検索し、さらにヒット構造を編集し再検索を行うなど、構造検索と分子設計をシームレスに行えます。
title:{フィンガープリント/QSAR/QSPR}
フィンガプリントによるフィルタリング
フィンガープリントとは、分子を部分構造の有無や立体構造的な特徴から算出されるハッシュ値のことです。MOEで利用可能なフィンガープリントには、部分構造、ファーマコフォア、分子形状に基づくものがあり、開発環境を用いてユーザー独自のフィンガープリントも追加できます。MOEではフィンガープリント間の類似度を算出して、大量の化合物群から類似分子だけに高速に絞り込めます。
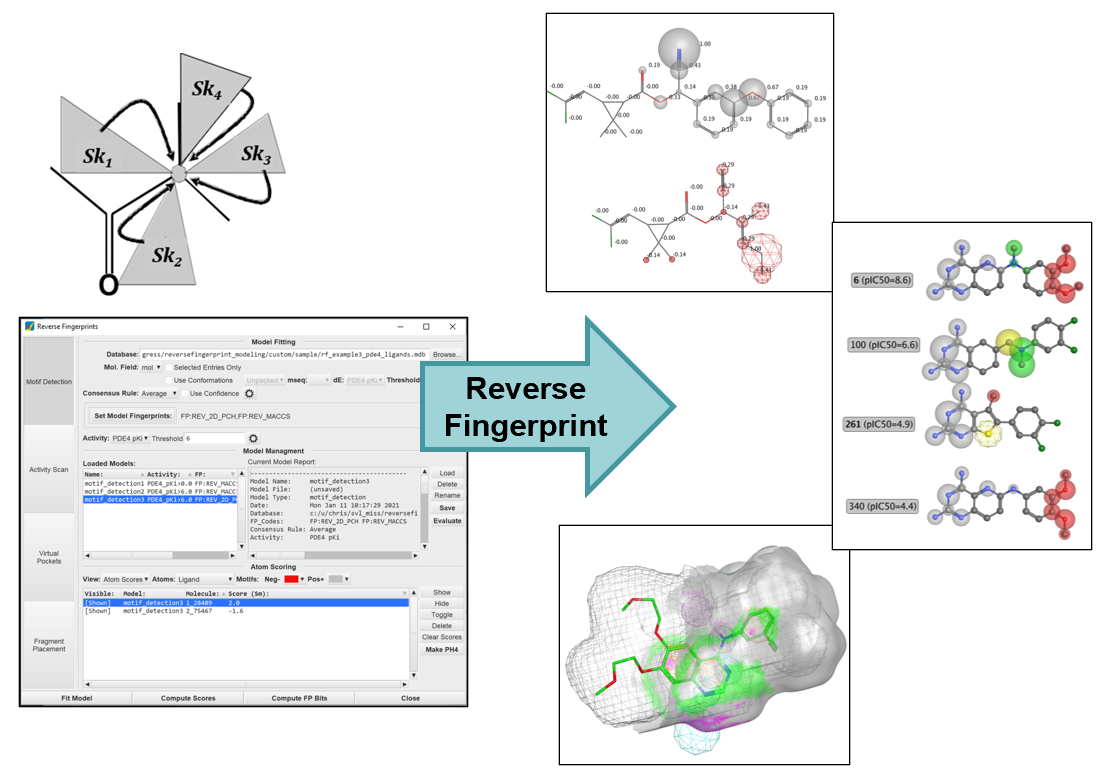
また、開発元が提供するアドオンプログラムであるReverse Fingerprint Modelingを用いることで、フィンガープリントから活性/不活性である確率と相関するスコアを求めることができ、バーチャルスクリーニングにおける化合物の順位付けに使用できます。このプログラムは、化合物のさまざまな分子フィンガープリントと活性値の情報を用いて、活性化合物に共通 の部分構造や活性 / 不活性に寄与する部分構造を特定したり、ファーマコフォアモデルを構築 / 評価したりできます。
QSAR/QSPRによるフィルタリング
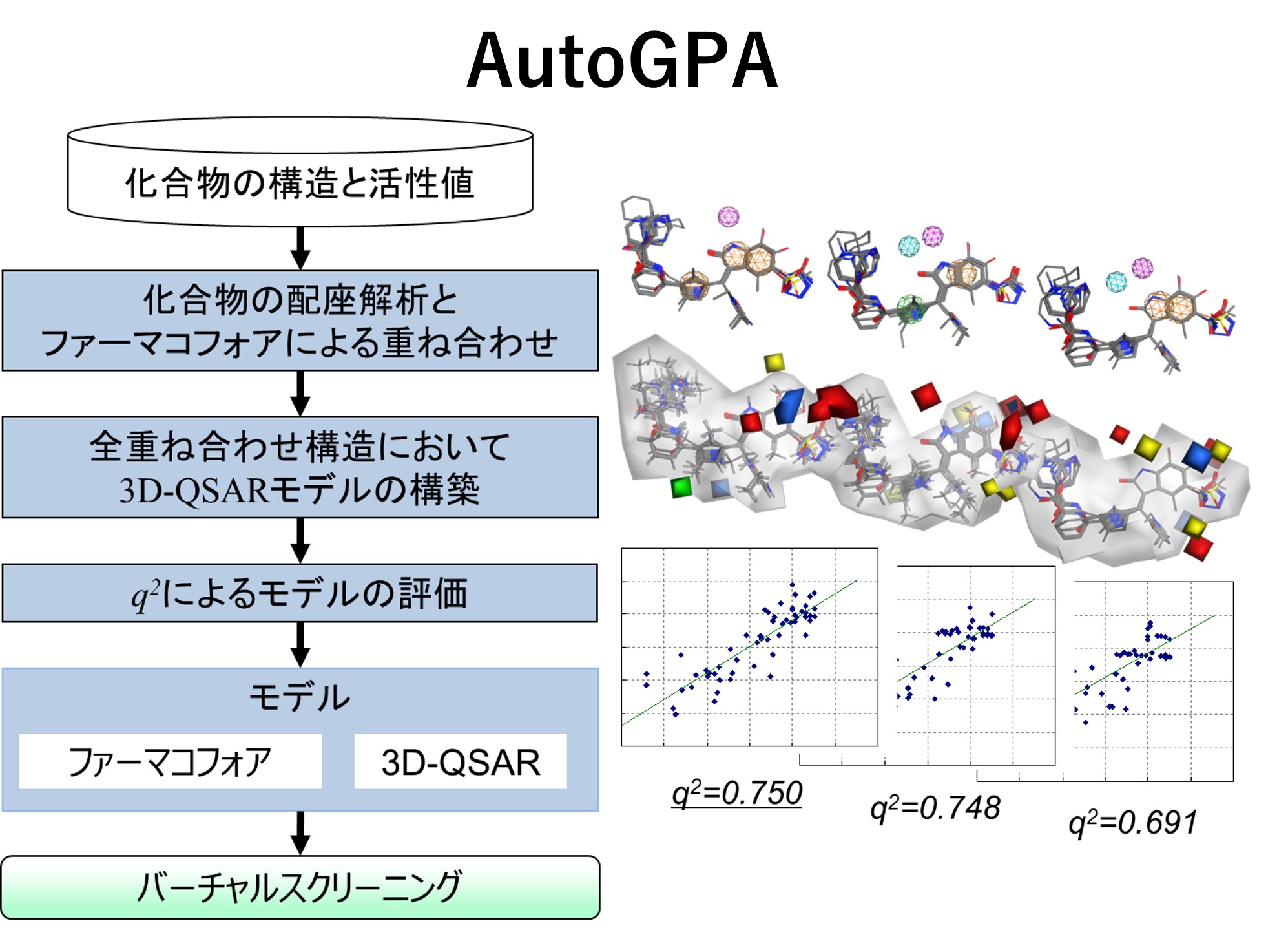
QSARは、化合物の置換基構造や物理化学的性質の違いと活性(受容体や酵素への結合活性、医薬品としての作用、毒性など)の強弱の間に認められる統計学的な関係性のことです(活性ではなく、物性の場合には、QSPRと呼ばれます)。MOEは、活性値や物性値を目的変数とし、分子構造から求められた分子記述子の値を説明変数として回帰することで活性(物性)予測モデルを構築できます。バーチャルスクリーニングにおいては、このモデルを用いて、例えば活性予測値が高い化合物群のみに絞り込めます。目的変数がpKi値のような連続値の場合、Partial Least Square Regression法あるいはPrincipal Component Regression法を用いて線形モデルを構築できます。また、目的変数が活性の有無(1/0)やクラス(A/B/C)等の離散値の場合、ベイズの定理に基づくBinary QSARや決定木を用いて非線形モデルを構築できます。また、開発環境を使用することで、別のモデル化手法をMOEに実装できます。当社が開発した自動QSARモデル構築ツールAutoQuaSARや自動3D-QSARモデル構築ツールAutoGPAは、MDB内に登録した分子構造と活性値から妥当なQSARモデルを自動的に構築します。
title:{ファーマコフォア}
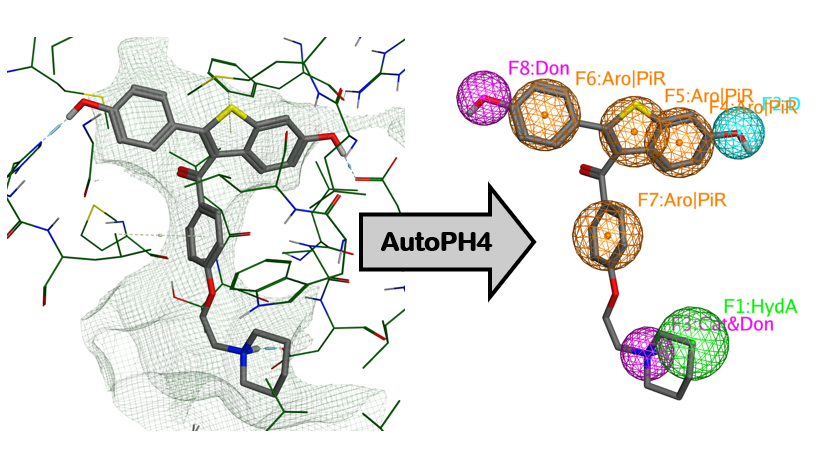
化合物を、部分構造や受容体との認識に関わる化学的特性(フィーチャー)の三次元配置でモデル化したものをファーマコフォアモデルと呼びます。ファーマコフォアモデルを用いることで、既存構造にとらわれずに、受容体との重要な相互作用が形成可能な化合物を三次元構造も含めて、大量の化合物群の中から高速かつ精度よく見出せます。MOEでは、ファーマコフォアモデルは、複合体構造から重要な相互作用を観察しながら構築したり、化合物群から共通するフィーチャーを抽出することで構築したりできます。ファーマコフォアモデルの詳細については、LBDDのページをご参照ください。また、開発元が提供するアドオンプログラムであるAutoPH4を使用すれば、複合体あるいは受容体の活性部位の構造のみから自動的に妥当なファーマコフォアモデルを構築できます。AutoPH4は論文として発表されており、他のファーマコフォアベースのバーチャルスクリーニングと比較して高い精度を達成しています。
title:{ドッキング}
ドッキングシミュレーションは、化合物のスクリーニングや最適な結合様式を評価するためにSBDDにおいては最も利用されるアプリケーションの一つです。多くの場合は、スクリーニングを行う目的でタンパク質の配座を固定した状態にして利用されますが、MOEのドッキングシミュレーションでは、誘導適合(induced fit)や共有結合ドッキング(covalent docking)にも対応しています。また、研究者の作業仮説を取り入れるための多数のオプションも用意されています。ファーマコフォアモデルや、X線結晶構造解析における電子密度分布を束縛条件としたドッキングシミュレーションも可能です。
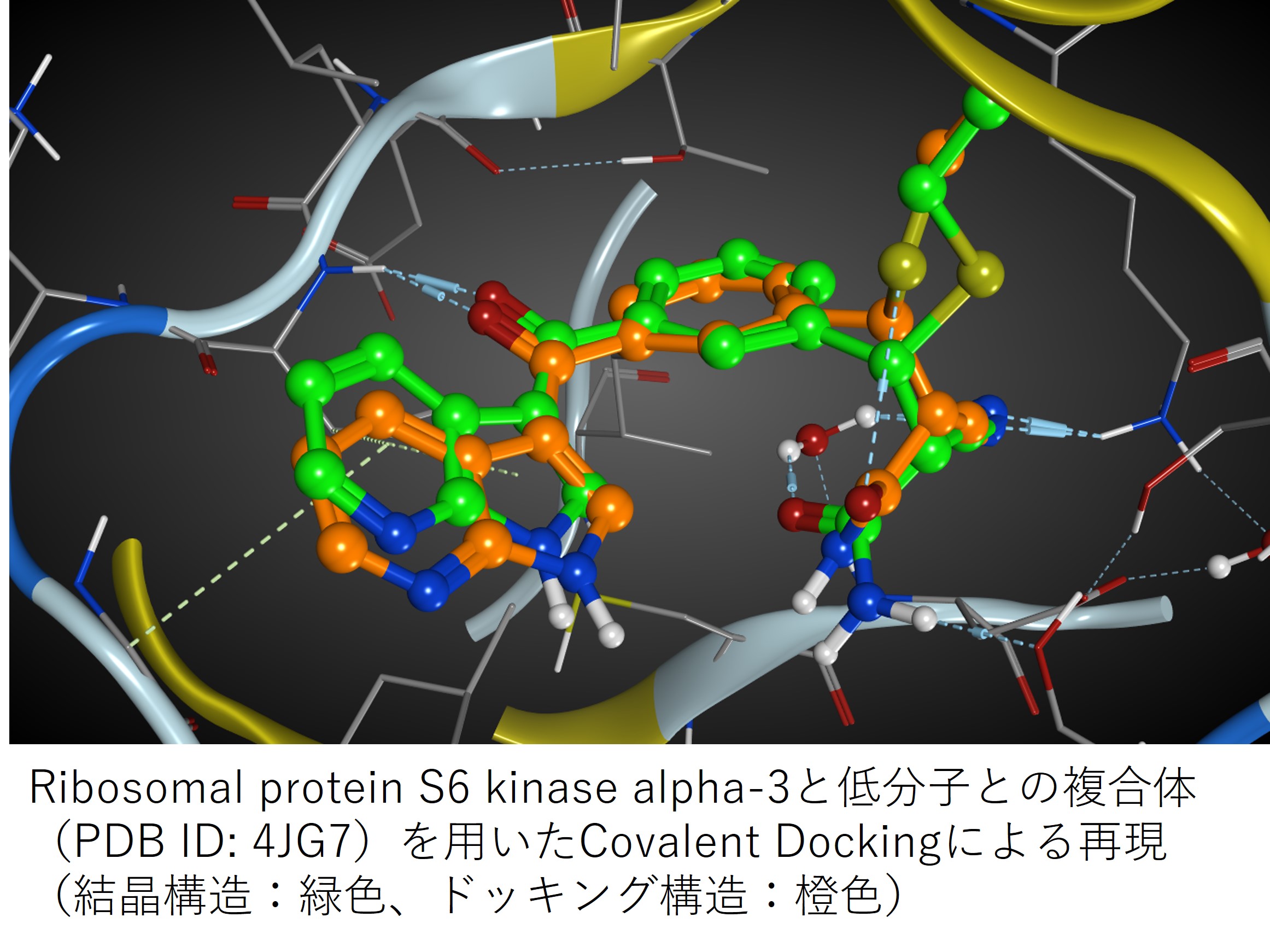
当社では、MOEの開発環境を用いて実装した、ドッキングシミュレーションプログラムASEDockを提供しています。低分子などの様々な リガンド候補化合物とタンパク質とのドッキング構造を高速かつ高精度に予測します。化合物のバーチャルスクリーニングに対応し、リガンドの部分構造重ね合わせや、タンパク質構造の誘導適合など数多くのオプション機能が搭載されています。
}:tab