


Molpro
(量子化学計算ソフトウェア)
tab:{
title:{概要}
概要
効率的な電子相関の取り扱い
Molproは、Werner教授(ドイツStuttgart大学)とKnowles教授(イギリスCardiff大学)を中心に開発された非経験的分子軌道計算ソフトウェアで、電子相関の精確かつ効率的な取り扱いに特長があります。Molproは、配置間相互作用法、クラスター展開法、多配置SCF法、多配置参照摂動法など電子相関の取り扱いに関する様々な計算機能を搭載しています。通常これらの方法は、分子サイズに対する計算コストが急激に増大するため小さな分子にのみ適用されてきましたが、Molproではintegral direct local electron correlation methods (1)を採用し、より大きな分子を取り扱うことができます。
(1) M. Schutz, Mol. Phys., 96, 719 (1999).
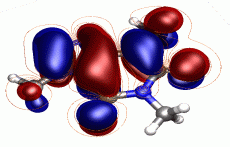
高精度な励起状態計算
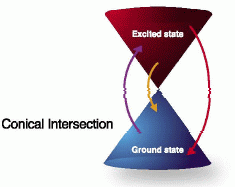
多配置波動関数を用いた計算手法に強みを持つMolproは、励起状態の取り扱いに優れたソフトウェアであるといえます。精確な励起状態波動関数を計算できますので、励起エネルギーの見積もりや状態間の円錐交差(コニカルインターセクション)の探索など、励起状態の研究に適しています。
パンフレット

ニュースレター
MOLSISニュースレターに掲載した記事を電子ブックで閲覧できます。
開発元ウェブサイト
Molproウェブサイト をあわせてご覧ください。
Molproの 更新情報 も確認できます。
title:{機能}
機能詳細
計算方法
原子価結合法: CASVB
Hartree-Fock法: RHF、UHF、ROHF、DF-HF
配置間相互作用法: CISD(T)、QCISD(T)、LQCISD(T)、DF-LQCISD(T)、FullCI, etc.
Møller-Plesset摂動法: MP2、MP3、MP4、LMP4、DF-LMP4、DF-LMP2-F12、DF-LMP2-R12, etc.
クラスター展開法: CCSD(T)、BCCD、LCCSD(T)、DF-LCCSD(T)、EOM-CCSD、CCSD(T)-F12, etc.
多配置SCF法: CASSCF、RASSCF
多参照Cl法: MRCI
多参照摂動法: MRPT2、MRPT3
多配置参照摂動法: CIPT2
密度汎関数法: LDA、GGA(Beck88、PW91、PBE、etc.)、Hybrid(B3LYP、BHLYP、etc.)
相対論効果: Scalar(DKH、Cowan-Griffin correction)、Spin-Orbit(BP operator)、ECP
溶媒効果: COSMO
外場の設定: 点電荷、静電場
構造予測・反応解析
構造最適化計算: Rotational Function、GDIIS、Quadratic Steepest Descent、Augmented Hessian
遷移状態探索: Rotational Function、GDIIS、Quadratic Steepest Descent
反応経路: QSDPATH
プロパティ計算
熱力学物性: エンタルピー、エントロピー、分配関数、モル比熱など
lRスペクトル
多重極子モーメント
励起エネルギーと強度
NBO解析、NPA解析
解析機能
MCSCF波動関数を用いたnon-adiabatic coupling matrix elements (1) の算出
CASSCF波動関数を用いた原子価軌道解析
MCSCF/MRCI波動関数を用いた一電子遷移特性
MCSCF波動関数を用いた二電子遷移特性
局在化軌道の出力
Distributed Multipole Analysis (2) による密度行列解析
非調和項を考慮した振動解析
(1) H. J. Werner, B. Follmeg, M. H. Alexander, J. Chem. Phys. 89, 3139 (1988).
T. Pacher, L. S. Cederbaum, H. Köppel, J. Chem. Phys. 89, 7367 (1988).
(2) A. J. Stone, Chem. Phys. Letters 83, 233 (1981).
title:{動作環境}
動作環境
OS
Linux 64 bit
macOS 64 bit
CPU
Intel/AMD x86_64
Apple Silicon
}:tab