

アプリケーション
SBDD 統合ツール SeeSAR 
SeeSARは、滑らかで美しい3Dモデルで分子間の親和力とリガンドの物性値を可視化します。SeeSARは、バイエル社、ハンブルグ大学、そしてBioSolveIT社で共同で開発された “Hydeテクノロジー”1),2),3)を搭載しており、原子単位での自由エネルギーを即座に算出します。
自由エネルギー寄与による原子の色付け、原子の存在しない空間の検出、ねじれ角の妥当性の検証、結晶構造の不自然な部分のチェックなどを行えます。SeeSARは、メディシナルケミストの日々の研究を支援する新しいリード最適化ツールです。
1) Reulecke, I. et al. Towards an Integrated Description of Hydrogen Bonding and Dehydration: Decreasing False Positives in Virtual Screening with the HYDE Scoring Function.
ChemMedChem, 2008,3, 885-897.
2) Schneider, N. et al. A consistent description of HYdrogen bond and DEhydration energies in protein-ligand complexes: methods behind the HYDE scoring function J Comput Aided Mol Des, 2013,27(1), 15-29.
3) http://www.biosolveit.de/SeeSAR/science.html
![]()
速くて、わかり易く、美しい これまでに類の無いリード最適化ツール
SeeSARは複雑な操作は不要で、簡単な操作だけでリガンド候補構造のリード最適化が可能です。
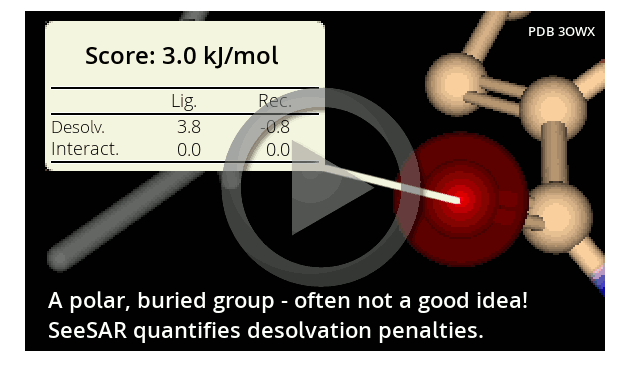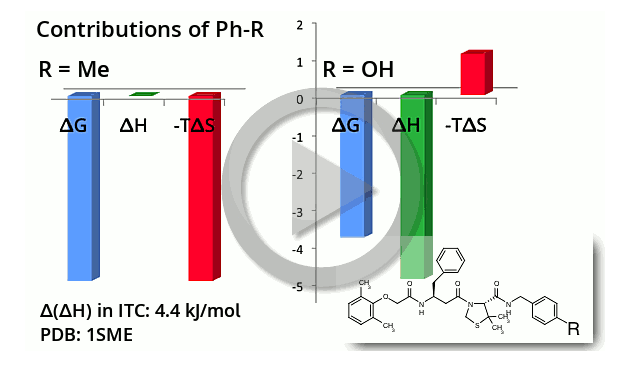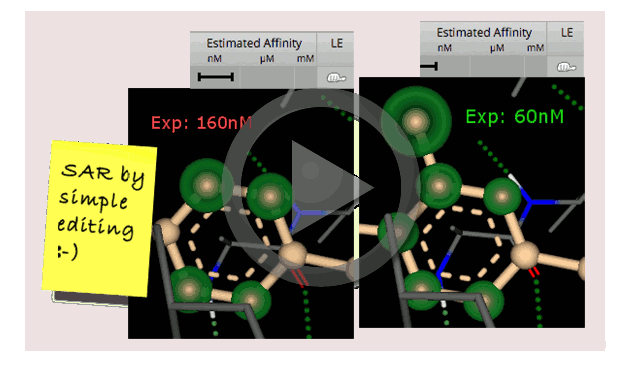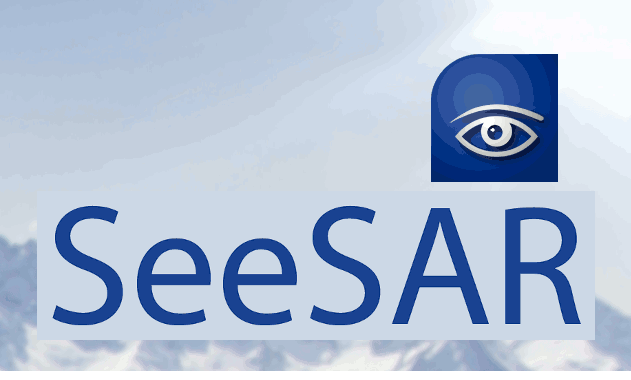
まず、SeeSARにタンパク質とリガンドの複合体構造を読み込むと、リガンド原子上に緑(ΔGが負の値)と赤(ΔGが正の値)の球が表示されます。これがリガンド各原子の結合自由エネルギーを表しています。
このような表示を行うことで、各原子が受容体との親和性にどのように寄与しているかを一目で認識することができます。SeeSARでは、この赤と緑の球の表示をビジュアルΔGと呼んでいます。
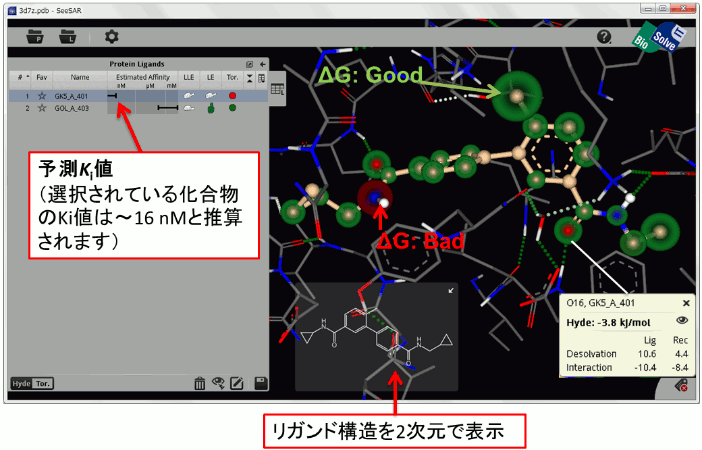
[PDB code: 3D7Z]
 ビジュアルΔG
ビジュアルΔG
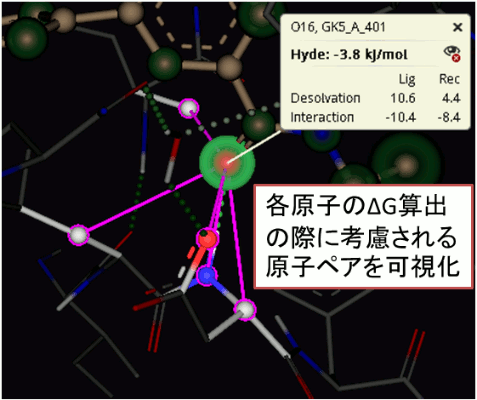
ΔGは、各原子におけるリガンド−受容体間の脱水和エネルギー(Desolvation)と水素結合エネルギー(Interaction)の合算値として評価されます。VisualΔGを評価に用いることで、例えば疎水性相互作用を視覚的かつ定量的に把握することができます。またΔGの算出に関与する原子を知ることで、ΔGの安定性の根拠を推測できます。
ねじれ角 (torsion angle)の解析
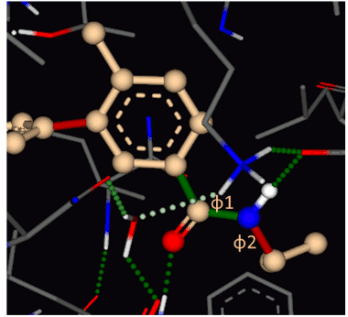
CSDデータベースの結晶構造から得た化合物のジオメトリーデータと比較してねじれ角の出現頻度を提示することで、原子間ねじれ角の妥当性の解析を行います。出現頻度は低(赤)、中(黄)、高(緑)で表現します。なお、この出力結果を得るためにCSDのデータベースは必要ありません。
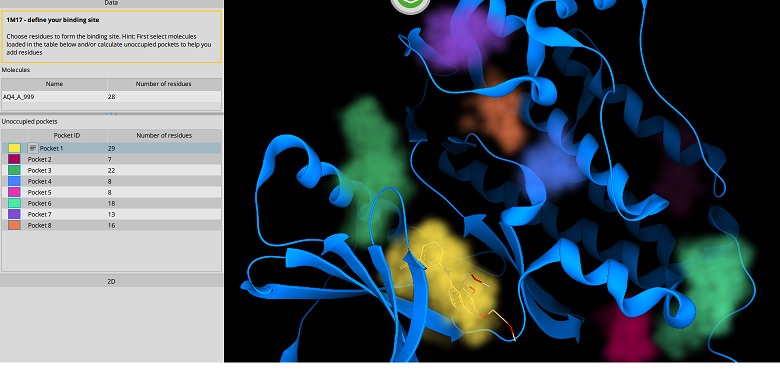
空間の検出
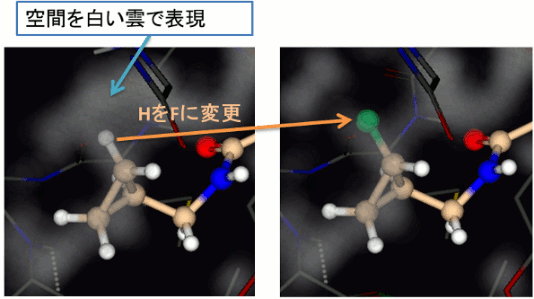
SeeSARはポケット内の空間を自動的に検出します。この隙間を利用してリガンドの改変を行うことで、より受容体と適合する候補構造を設計することが可能です。
水素原子付加
ProToss4)によるプロトン/脱プロトン化、OH、NH の回転、互変異性体の選択、側鎖アミドのフリップを考慮した精度の高い水素原子付加を世界最高水準のスピードで実行します。
4) Bietz, S.; Urbaczek, S.; Schulz, B.; Rarey, M., Protoss: a holistic approach to predict tautomers and protonation states in protein-ligand complexes, J. Cheminform., 2014, 6:12.
リガンド結合サイトの検出
DoGSiteScorer アルゴリズムを用いて、ポケットの大きさ、形状、化学的特徴に基づいてリガンド結合サイトを検出します。リガンドとの共結晶構造を持たない分子系においても、結合ポケットを検出しドッキングシミュレーションなどの解析が可能です。
使いやすく、高速な計算
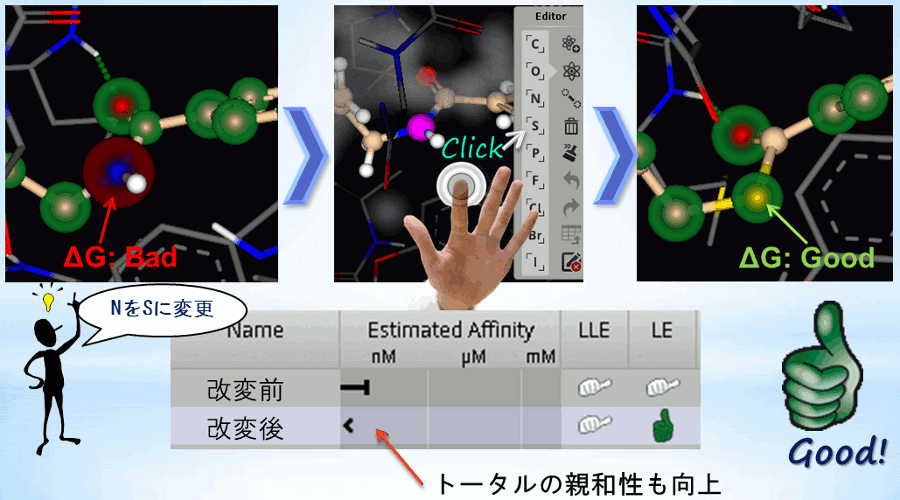
SeeSARは極めて直感的に簡単なマウス操作だけで分子設計が可能です。分子構造ファイルの入力はGUI左上にある「P」、「L」ボタンから読み込みます。「P」ボタンを押して、受容体-リガンド複合体構造を入力すれば、即座に結合親和性の
予測値やLE (ligand efficiency), LLE(lipophilic ligand efficiency)等が計算され、その計算値をリスト表示します。リストからリガンドを選択することで結合自由エネルギー計算も即座に行われ、研究者がイメージしたものをすぐに具現化できます。たとえば、リガンドの親和力を向上させるためには下図のような操作を行います。
さらに「L」ボタンからSDファイルを入力することで、異なるリガンドや異なる配座をリストに加えることができ、それらの結合親和性や結合自由エネルギーを計算することができます。
また、「P」ボタンからリガンドを含まないタンパク質受容体のみを読み込ませることも可能です。
ドッキングシミュレーション
化合物のドッキングシミュレーションが可能です。ファーマコフォアを基準としたドッキングシミュレーションや、リガンド構造あるいは任意の部分構造をテンプレートとしたテンプレートドッキングが可能です。
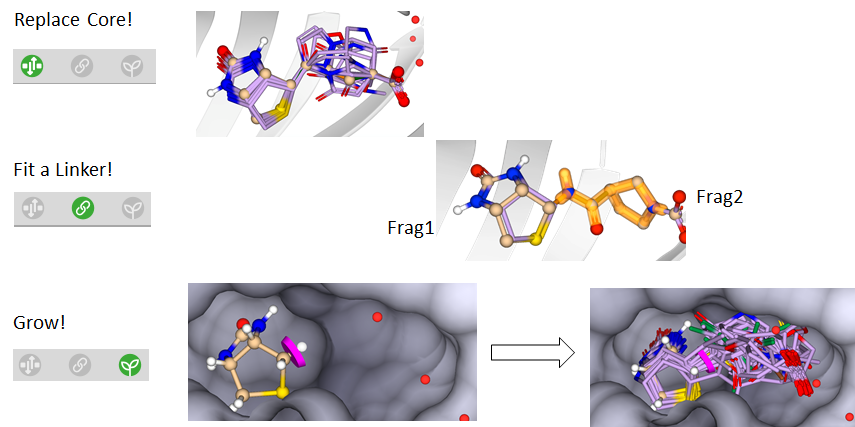
母核置換・フラグメント伸長・フラグメント連結
フラグメント構造を用いて新しいリガンドのデザインが可能です。母核置換は、リガンドの母核構造部分を選択し置き換えます。フラグメント付加は、フラグメントを伸長しポケットと適合するリガンドを出力します。フラグメント連結は、複数のフラグメントを繋げるためのリンカーを探索します。
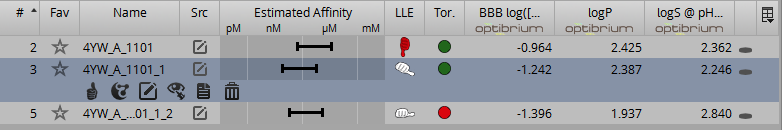
化合物のADME特性計算
新規リガンド構造の探索において、活性値だけでなく
ADME特性を確認しながら分子設計を行うことは創薬研究の
効率化につながります。
SeeSARで読み込んでいる化合物データに対して、
Optibrium社のStarDropのADME特性が計算可能です。
計算可能なADME特性は以下です。
– logP (Octanol/Water)
– logD (Octanol/Buffer at pH7.4)
– Aqueous Solubility, Solubility at pH 7.4
– Cytochrome P450 Affinities (CYP2C9, CYP2D6)
– Blood Brain Barrier Penetration
– Human Intestinal Absorption
– P-glycoprotein substrate
– Plasma Protein Binding
ADME特性は、受容体構造のない化合物データだけでも評価可能です。
SeeSARを用いた計算例
置換基を変化させた際の結合親和性の見積もり
置換基を変えることで、リガンドの結合親和性が変化することがあります。ここでは化合物の一部の置換基を改変した場合の、リガンドの結合親和性の実験値とSeeSARによる予測値の比較を行います。
下の図表では化合物にメチル基を付加し、結合親和性が良くなる例です。このようにメチル基によって、結合親和性が改善するものを「Magic Methyl」と呼んでいます。
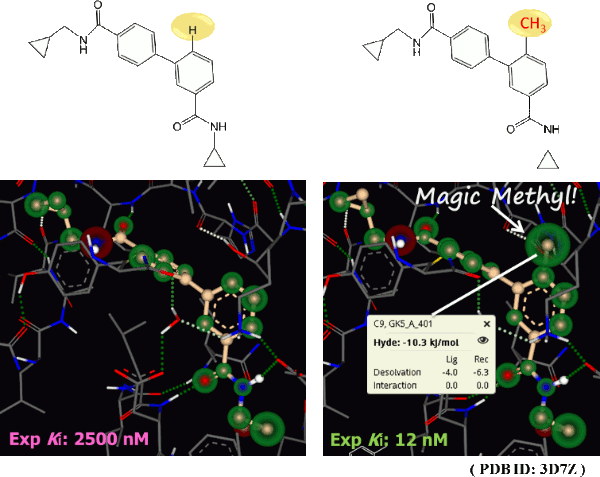
上の図では、メチル基の炭素原子上に大きな緑色の球が描かれています。つまり、ΔGが負となり結合親和性向上に寄与していることがわかります。
下の図は、メチル基以外にも様々な置換基に変更した場合の実験値5)と計算値との比較です。
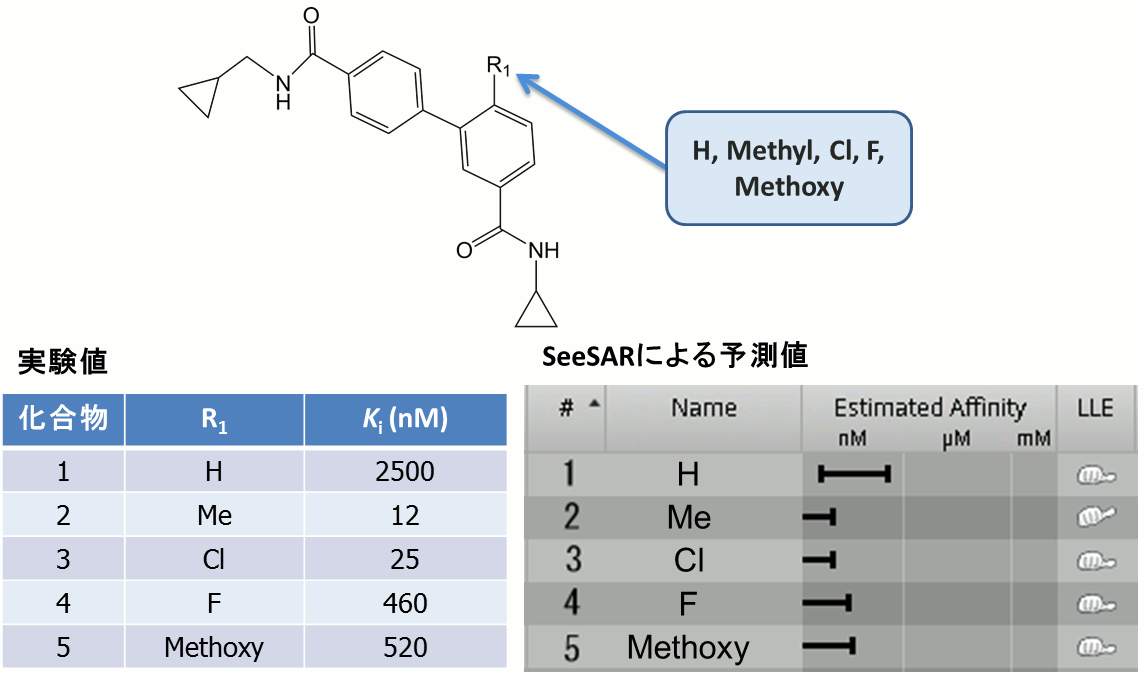
上の2つの表を比較しますと、SeeSARによる計算値と実験値との相関が高いことがわかります。
5) Angell,R. et al. Biphenyl amide p38 kinase inhibitors 3: Improvement of cellular and in vivo activity, Bioorg. Med. Chem. Lett., 2008, 18, 4428-4432
ドッキング結果のリスコアリング – 再ドッキング(Redocking)を例に –
PDBの複合体構造を用いて、リガンドを再ドッキングすることで、スコアリングの妥当性を検証します。ここではヒトアデノシンA2A受容体(PDBコード:3EML)の既存リガンドの再ドッキング計算を行いSeeSARで結合親和性の推算を行いました。
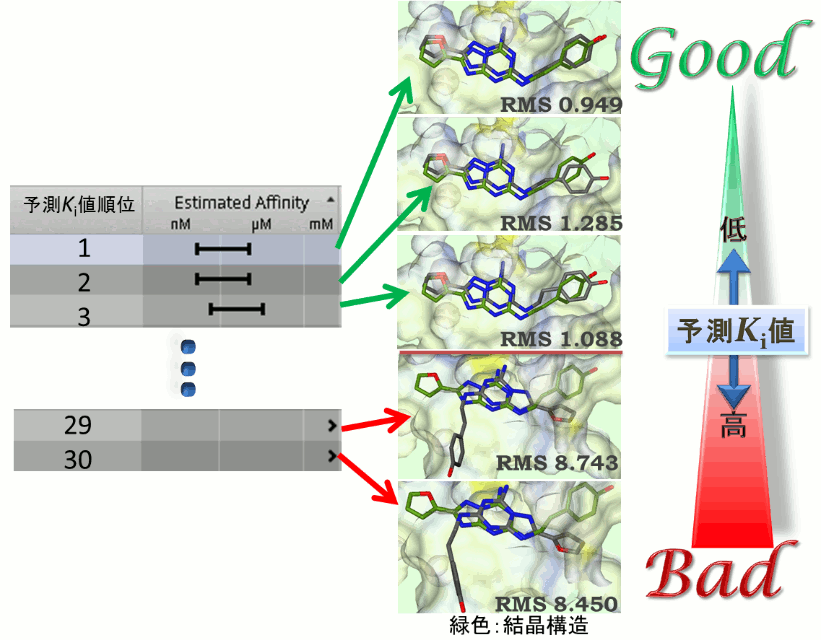
上の図では、予測したKi値が大きいほどPDBの結晶構造に含まれるリガンドとのずれが大きくなる傾向が見られました。
下の図は、ここで得られたデータをもとにROC解析した結果です。ここでは、オリジナルリガンドからのRMSが2以下のものをpositiveと定義しました。
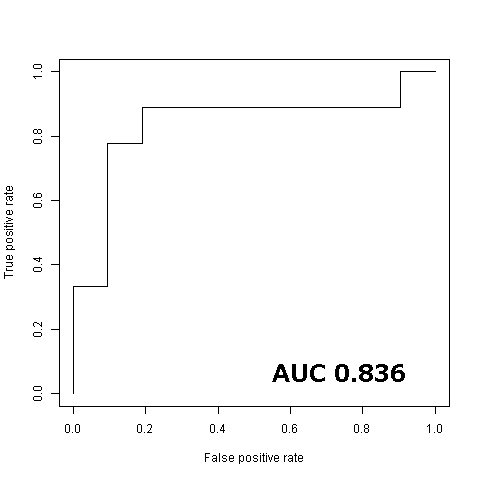
上記の例ではAUCが0.836となり、SeeSARによって予測したKi値はRMSの低いコンフォメーションを有意に分離することができることがわかりました。
ドッキング結果のリスコアリング – DUDのドッキングベンチマークセット6)による検証 –
DUDのドッキングベンチマークセットを用いて、既知リガンドとデコイデータとを使ってドッキングコンフォメーションを得て、SeeSARのスコアでそれらの差異を見出すことで、スコアリングの妥当性を検証します。
ここではFlexXを用いてレチノイドX受容体α(PDBコード:1MVC)に対して既知リガンドおよびデコイ化合物のドッキング計算を行い、ドッキングコンフォメーションに対してSeeSARで結合親和性の推算を行いました。
計算の結果、デコイ化合物と既知のリガンドとを有意に分離することができました。
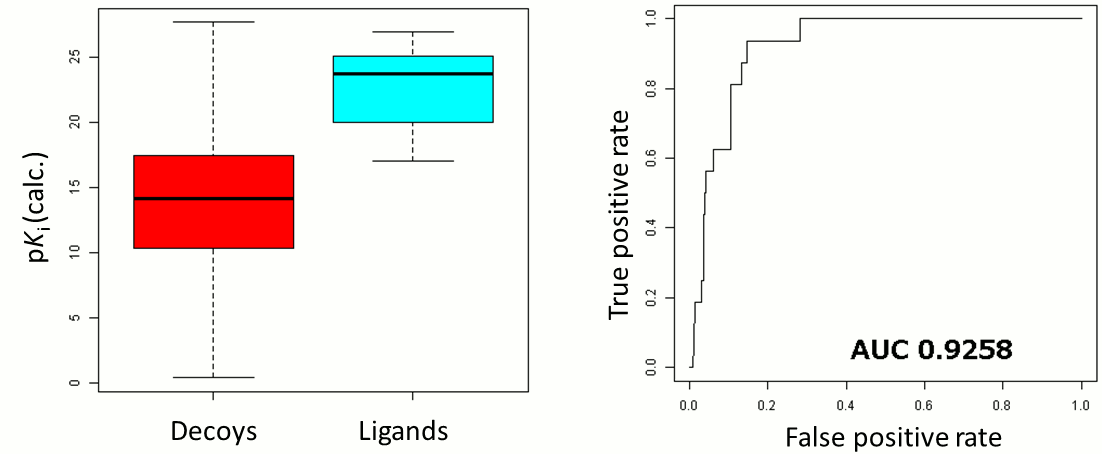
6) Huang,N. et al. Benchmarking Sets for Molecular Docking, J. Med. Chem., 2006, 49(23), 6789-6801
