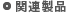製品構成
DFTB
 密度汎関数法に基づくタイトバインディング計算プログラム
密度汎関数法に基づくタイトバインディング計算プログラム
 DFTB(Density Functional based Tight-Binding)は、密度汎関数法に基づくタイトバインディング計算プログラムです。DFTBでは、電子間相互作用の積分計算をパラメータ化することで高速かつ精度の高い計算が実現されており、通常の密度汎関数法計算では取り扱えない大規模な系にも適用することが可能です。DFTBパラメータは、Quasinano*プロジェクトによって開発されたパラメータを利用可能で、周期律表のほとんどの元素をサポートしています。DFTBのハミルトニアンでは、荷電系の正確な取り扱いを可能にするためのSelf-consistent charge (SCC-DFTB)法の搭載や、長距離相互作用を記述するための分散力補正などがなされています。
DFTB(Density Functional based Tight-Binding)は、密度汎関数法に基づくタイトバインディング計算プログラムです。DFTBでは、電子間相互作用の積分計算をパラメータ化することで高速かつ精度の高い計算が実現されており、通常の密度汎関数法計算では取り扱えない大規模な系にも適用することが可能です。DFTBパラメータは、Quasinano*プロジェクトによって開発されたパラメータを利用可能で、周期律表のほとんどの元素をサポートしています。DFTBのハミルトニアンでは、荷電系の正確な取り扱いを可能にするためのSelf-consistent charge (SCC-DFTB)法の搭載や、長距離相互作用を記述するための分散力補正などがなされています。DFTBを利用することで、大規模な系に対しても長時間のシミュレーションを実行することがパソコンレベルの計算機で実現できます。DFTBは周期系だけでなく孤立分子系にも対応しています。より高精度な計算(密度汎関数法など)を実行する前の簡易構造最適化計算プログラムとしてDFTBを利用することもできます。SCM社製ソフトウェアでは密度汎関数プログラムとしてADFやBANDが利用可能で、これらのプログラムと連携した取り扱いが可能です。
*) Quasinanoプロジェクトは、独Jacobs大学と蘭SCM社の共同研究による取り組みで、軽元素だけでなく遷移金属も含むナノサイズの系(10万原子程度)を高速かつ精確に扱える手法の開発を目指したプロジェクトです。
DFTBの主な機能
- 計算方法
-
- Self-consistent charge (SCC-DFTB)法
- third-order self-consistent charge (DFTB3) 法: DFTB.orgパラメータのみ対応
- 分散力補正(D3, D3-BJ, UFF)
- 時間依存(TD-DFTB)法
- 非平衡グリーン関数(NEGF-DFTB)法
- DFTBパラメータ
-
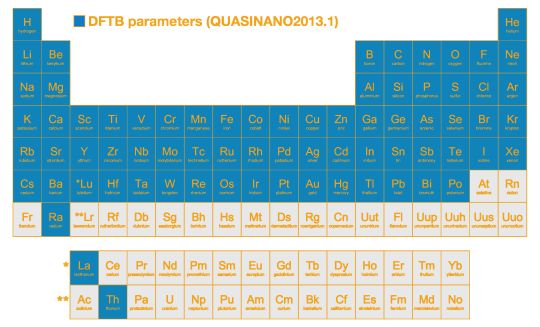
- QUASINANO 2013.011 (周期律表の多くの元素に対応:下図参照)
- Dresden set (Al-O-P-C-H, Al-Si-O-H 等)
- DFTB.org (H-C-N-O-S-P, Si-F-O-N-C-H 等):非営利団体は無償利用可
1) M. Wahiduzzaman et al., J. Chem. Theory Comput. 9, 4006(2013).
Quasinano2013.1では、斥力ポテンシャルは含まれていないため、
力の計算を要する構造最適化や分子動力学計算には利用できません。