


MOE - 分子シミュレーション
tab:{
title:{概要}
分子シミュレーションとは、計算機を用いて物質科学全般の現象を探るための方法論です。分子のポテンシャルエネルギーや最安定構造など、物質の特性を分子レベルで解明することができます。
title:{分子構造の前処理}
水素原子付加状態の最適化(Protonate3D)
X線結晶構造解析による生体高分子の立体構造データの多くには、水素原子の位置情報が含まれていません。ポテンシャルエネルギーは水素原子の有無や位置、イオン化状態によって大きく変化するため、生体高分子のシミュレーションではまず水素原子の位置を正確に予測することが重要です。Protonate3Dは、立体構造の取り得る水素原子付加状態から最も安定な状態を探索する機能です。温度、pH、塩濃度を考慮して、タンパク質や結合している化合物のイオン化状態、互変異性体、水素原子の位置、側鎖のフリップ状態を最適化することで最もエネルギーが安定となる水素原子付加状態を予測します。取り得る膨大な状態の組み合わせから高速に最適解を求めるUnary Quadratic Optimization (UQO)アルゴリズムを搭載しています。
Labute, P. Protonate3D: Assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins: Struct., Funct., Bioinf. 2009, 75, 187–205.
低分子のイオン化状態の予測(Protomers)
指定のpHにおける低分子のイオン化状態や互変異性体の存在比率を予測します。分子記述子と拡張Hückel法により得られたパラメーターを利用した独自のpKa予測モデルを用いています。
タンパク質立体構造の問題点の確認と修正(Structure Preparation)
タンパク質立体構造データに含まれる分子シミュレーションを行う上での問題点を検出し、自動的に修正します。欠損している原子座標の補完やキャッピング、ジスルフィド結合の再構築、残基名の修正、部分電荷計算、水素付加状態の最適化、原子間衝突/シス型ペプチド結合の検出などが行えます。
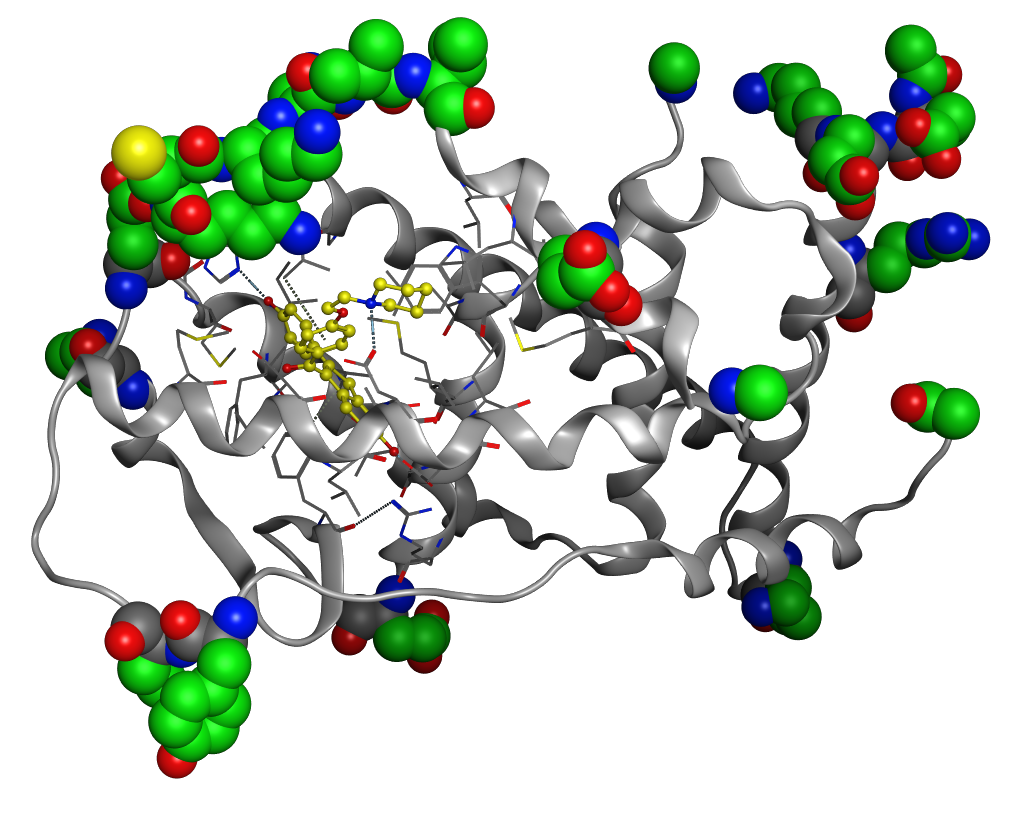
PDB ID: 1ERRにおけるStructure Preparationでの構造補完。緑の部分がStructure Preparationで修正した構造。
分子構造の自動前処理(QuickPrep)
タンパク質構造や、タンパク質ーリガンド複合体構造に対して適切な前処理を自動的に実行することができます。計算化学者が一般的に行う処理を自動化し、タンパク質立体構造の問題点の修正(Structure Preparation)、相互作用していない水分子の削除、水素原子付加状態の最適化(Protonate3D)、リガンド結合部位の構造最適化計算を連続で実行します。ユーザーはこの前処理を行うだけで、分子シミュレーションやSBDDをシームレスに実施できます。
title:{分子力学計算}
ポテンシャルエネルギー計算
分子系のポテンシャルエネルギーを推算します。これにより、分子の配座の違いによるポテンシャルエネルギーの差や、リガンド-受容体複合体など分子間の相互作用エネルギーを計算できます。計算に使用する分子力場として、拡張Hückel法に基づいた原子タイプ非依存的な力場Amber:EHT/Amber:EHToを搭載しており、高精度なエネルギー計算が可能です。溶媒効果として、距離依存誘電率や反応場、一般化ボルンモデル(GBVI)を利用できます。
構造最適化計算
初期構造から、よりエネルギーの小さい構造を算出し安定な分子構造を求める方法です。平衡状態にある分子配座の一つを短時間で求めることができます。化合物の2次元構造から3次元構造への変換にも使用できます。並列計算に対応しているため、タンパク質など高分子を含む系でも効率良く高速に計算を行えます。必要に応じて配座や一部の原子座標の固定や、原子間距離・角度・二面角の拘束を設定できます
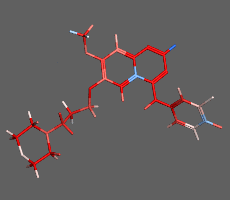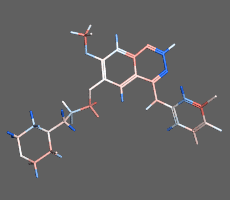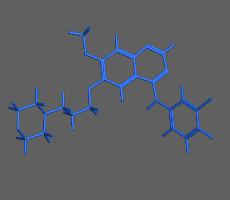
構造最適化計算(エネルギー極小化計算)の例:構造ひずみの大きい(赤)初期構造から安定な極小化構造(青)を算出
title:{分子動力学計算}
分子動力学計算
分子動力学法は、運動方程式に従って原子や分子集団の時間発展を追跡するシミュレーション方法です。これにより、熱運動によるタンパク質の揺らぎや分子の拡散速度などの動的な振る舞いを表現することができます。 熱力学的な系の条件として、温度や圧力、体積などを一定もしくは経時変化するよう指定できます。溶媒の配置と周期境界条件の設定を行うインターフェースから容易に系を構築して分子動力学計算を実行できます。計算エンジンとしてMOEの他に、NAMD*やAMBER*を利用することができます。これらのソフトウェアではマルチコア、クラスター、GPUを利用した高速な計算ができます。AMBERの熱力学積分法による結合自由エネルギーの推算もできます。

HIVプロテアーゼと阻害剤の複合体に周期境界条件を設定した分子動力学計算の初期構造
*別途入手が必要です。
title:{低分子の解析}
配座解析
分子の取り得る様々な安定配座を発生し、最安定構造を検索する方法です。MOEでは網羅的に二面角を回転させて探索するSystematic法や、ランダムに探索するStochastic法、低振動モード解析による初速度の重み付けをした分子動力学法による熱運動からサンプリングするLowModeMD法が搭載されています。目的の分子の種類にあわせて配座解析手法を選択できます。特にLowModeMDは、低分子、ペプチド、環状構造、タンパク質のループ部分の配座解析など効率的に配座のサンプリングを行えます
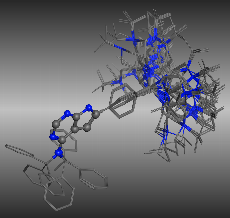
配座解析の結果例:Ball&Stick表示部分で重ね合わせたもの
Torsion Profile
低分子における特定の結合二面角を段階的に回転させたときのエネルギー分布を解析する機能です。配座の柔軟性の評価、回転障壁の推定、好ましい二面角の特定に使用できます。MOEの分子力場だけでなく、量子化学計算ソフトウェア*を用いたエネルギー評価もできます。また、Mogul*(ナレッジベースの分子ジオメトリーのデータベース)を用いると、ケンブリッジ結晶構造データベースを基にした二面角のヒストグラムを合わせて出力できます。
Torsion Analyze
CSD 統計との整合性に基づいて、二面角の品質を動的に評価することができる機能です。品質を示す指標として各結合にスリーブが表示されます。
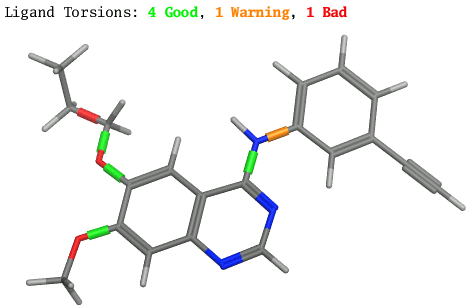
Torsion Analyzeによって各二面角の評価した分子
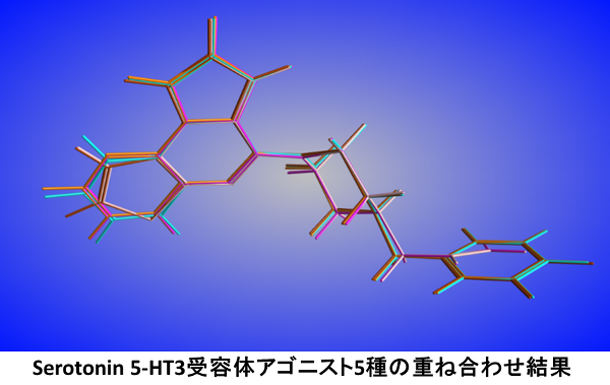
分子アライメント
Flexible Alignmentは、複数の低分子を自動で重ね合わせる機能です。各分子のもつ水素結合アクセプター/ドナーなどの特性がより良く重なるように配座を変化させたフレキシブルな重ね合わせを行います。分子配座や一部の原子座標を固定した重ね合わせ、テンプレート構造を使用した重ね合わせも可能です。得られた重ね合わせは特性の重なりとひずみエネルギーで評価されます。その他の機能として、基準の分子に対して、複数の手法(分子構造全体、任意のフラグメント、SMARTS、類似3原子、分子配座)に基づいてデータベースの分子を連続的に重ね合わせるMolecule Superposeがあります。
*別途入手が必要です。
title:{静電ポテンシャル解析}
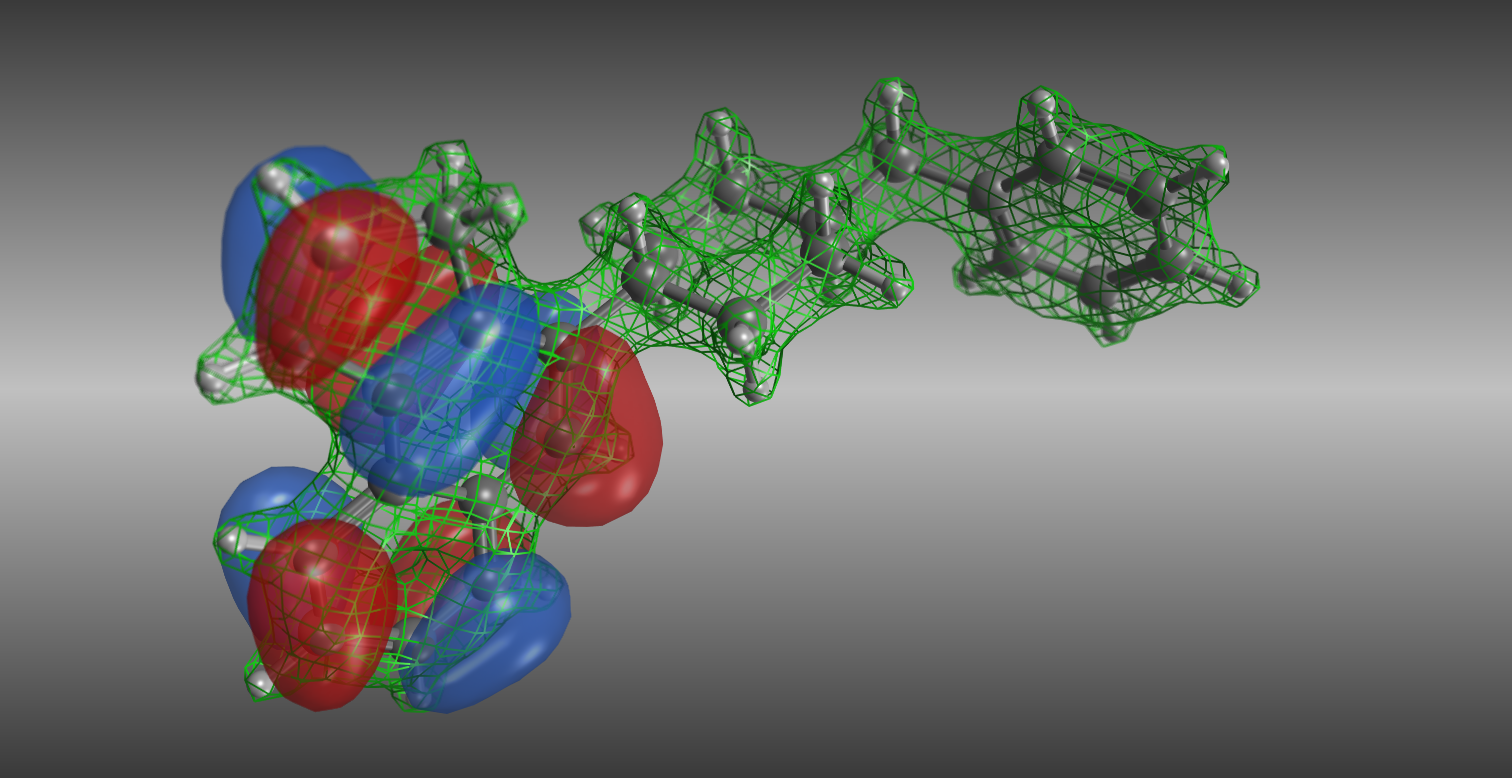
静電ポテンシャル解析
重ね合わせた化合物から、構造の「掛け合わせ」を行い、新規化合物を発生させます。複数の化合物で共通する結合位置を基準にして、置換基をお互いに入れ替えます。受容体構造がある場合は結合部位において構造最適化を行います。

title:{量子化学計算}
スペクトル解析(Spectral Analysis)
低分子の構造決定に有用なスペクトル解析機能です。量子化学計算ソフトウェアのGaussian*による核磁気共鳴(NMR)スペクトルと振動円二色性(VCD)スペクトルの計算結果の可視化、実験結果との比較ができ、構造異性体や光学異性体の構造決定に利用できます。NMR実験により得られたケミカルシフトやJ-カップリング、NOEデータを用いることで溶液中の分子配座の存在比を求めることができます。
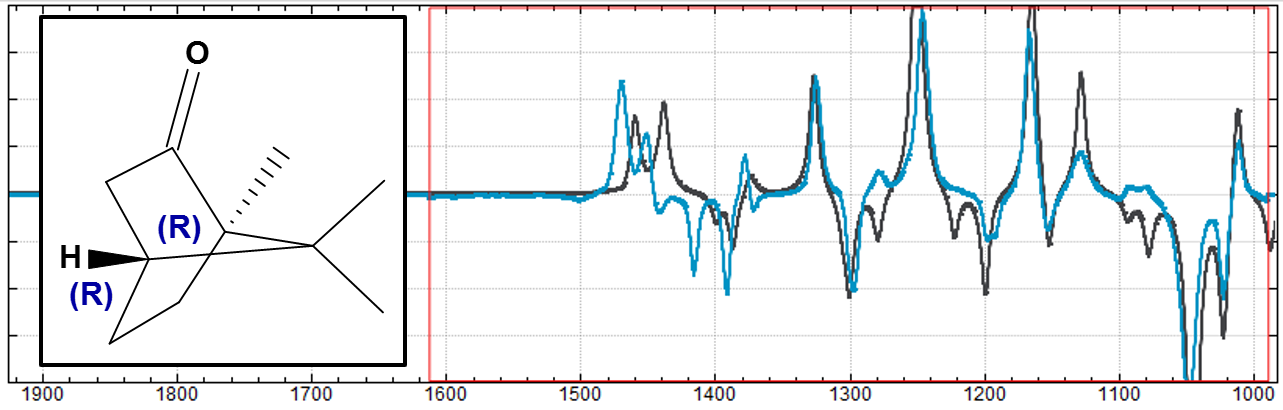
Camphorの構造式(左)とVCDスペクトル(右)。計算結果(黒)は、11.25cm-1の振動数シフト、0.98のスケール因子で実験結果(青)に調整。
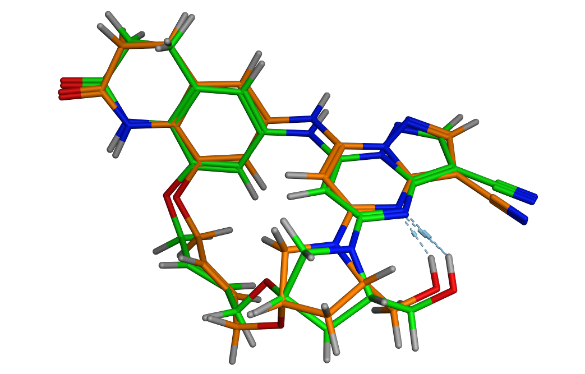
環状リガンドの溶液中での配座分布解析。黄橙: 溶液中のNOEの実験結果に近い配座、緑:結晶構造。
*別途入手が必要です。
}:tab